Bioimpacts. 15:31459.
doi: 10.34172/bi.31459
Review
CAR-T cell therapy as an approach for pediatric hematological malignancies in regenerative therapy: Current status and clinical outcomes
Xiaoman Guo Conceptualization, Supervision, Writing – original draft, Writing – review & editing, 1, * 
Xueyin Jiang Writing – original draft, 1
Juan Zhang Writing – original draft, 1
Author information:
1Department of Pediatrics, Yantaishan Hospital, No.10087, Keji Avenue, Laishan District, Yantai, 264003, Shandong Province, China
Abstract
Radiation therapy, chemotherapy, and surgery have been the standard cancer treatment approaches for many years. Even with these treatments, the majority of tumors still have a dismal prognosis. With complete remission rates ranging from 65% to 90% in the crucial CD19-CART trials, chimeric antigen receptor T-cell (CART) therapy has revolutionized the treatment paradigm for pediatric patients with relapsed/refractory B-cell acute lymphoblastic leukemia (B-ALL). Hematological tumors have responded well to CART. The first CART was authorized by the FDA in 2017 to treat B-ALL. The FDA authorized CART to treat B-cell lymphoma in October of that year. In recent years, research has focused on CART to increase and improve the therapeutic effect. New toxicity profiles and treatment constraints have also surfaced with this new medicine, calling for cooperative group trials, new management strategies, and toxicity consensus grading systems. The introduction of CART treatment for pediatric B-cell ALL will be the main topic of this article, along with previous and ongoing trials. We will also talk about CART therapy trials for various pediatric cancers. Safe procedures and close observation are essential since CART treatment has the potential to cause serious toxicities.
Keywords: Acute lymphoblastic leukemia, CART, CAR-T cell therapy, Clinical outcomes, Pediatric hematological malignancies
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
No funding source was required.
Introduction
One type of targeted immunotherapy that makes use of tumor-specific antigen recognition is chimeric antigen receptor T-cell (CART) treatment.1 Customized receptor known as chimeric antigen receptors (CARs) target particular antigens on cancerous cells.2 Apheresis is used to harvest a patient's own T cells, which are subsequently used in the production of CART. The T cells are genetically altered to express the CAR during this phase. The CART is prepared for patient infusion upon completion of manufacture. The therapy's objective is for the modified CART to identify and target the antigen, multiply, and endure in order to provide long-term disease monitoring.3
The basic idea of CART treatment is to genetically alter T lymphocytes so they can identify particular, distinct targets on tumor surfaces and produce cytotoxic effects.4,5 CART therapy has shown great success in treating solid tumors and a variety of hematologic malignancies.6 The landscape of pediatric relapsed B-cell acute lymphoblastic leukemia (ALL) treatment changed in April 2012. The first pediatric patient with relapsed B-cell ALL received anti-CD19 CART via targeted immunotherapy.7 Today, the patient is still in remission. Increased research and trials utilizing CART products for relapsed and refractory B-cell ALL (B-ALL), as well as other relapsed pediatric cancers such as T cell ALL, acute myeloid leukemia (AML), lymphoma, and solid tumors like neuroblastoma and brain tumors, have been spurred by this extraordinary success. Due to the favorable results associated with CART therapies for B-cell malignancies and other significant immunotherapy research, immunotherapy has emerged as the fourth treatment modality for cancer, alongside chemotherapy, radiation, and surgery.8
For the treatment of patients with multiple relapsed and/or refractory B-ALL, the majority of CART treatments studied in pediatrics have targeted B-cell antigens. Different CART constructions have been tried in a number of clinical trials with a range of patient groups, and each trial has shown remarkable rates of inducing remission. More information about AML and other T- and B-cell cancers is becoming available. In high-risk patients with refractory or multiple relapsing illness, CART treatment is still utilized as salvage therapy despite its great efficacy. To determine the best time to administer CART within the current therapy paradigm, clinical trials are still being conducted.9
CART technology
CART is composed of extracellular single-chain antibody fragments, transmembrane domains, and intracellular T cell signaling domains.10 T cell receptors exist as major histocompatibility complexes and antigen-peptide complexes and are dependent on antigens provided by antigen-presenting cells.11 An intracellular cascade reaction is triggered when the T cell receptor (TCR) and major histocompatibility complex (MHC) antigen peptide complex are combined. The first signal is produced by the phosphorylated TCR recruiting intracellular second messengers, and the second signal is produced by the costimulatory molecules on the surface of T cells (CD28, CD27, CD134, CD137, or ICOS) and the corresponding receptors on antigen-presenting cell (APC) (CD80, CD86, CD137L, or ICOSL). The extracellular antibody in CAR, in contrast to TCR-T, attaches to the appropriate tumor antigen, identifies it, and activates T cells in a way that is not MHC-based to provide anti-tumor effects.12 CARs can be divided into five generations based on how their intracellular signaling domain is organized, even though their basic modular structure hasn't changed since they were first created.
First-generation CART
The CD3 ζ-chain or FcεRIγ intracellular domain was the only costimulatory domain present in first-generation CARs. These complexes shared many characteristics with endogenous TCR, but they had a significant flaw: they were unable to generate enough interleukin (IL)-2. Exogenous IL-2 had to be added to first-generation CARs in order to guarantee an effective response because of their poor response. Additionally, research showed that these altered cells continued to exhibit poor cell proliferation and a brief in vivo lifespan, which further stimulated the creation of costimulatory domains.13,14
Second-generation CARs made an effort to address the issues brought on by traditional CART's short lifespan, poor cytokine production, and insufficient proliferation. They achieved this by utilizing dual signaling, which is known to promote robust T cell proliferation in natural systems.
Second-generation CART
Other cytoplasmic domains, including CD28, 4-1BB, or OX-40, were present in this new generation of CARs and may provide a secondary signal when they came into contact with a tumor antigen. According to preclinical and clinical research, the costimulatory signal's prolonged in vivo half-lives allowed it to enhance cytotoxicity, sustained response, and proliferation. Research also showed that these characteristics were significantly modulated by the costimulatory domain's composition. According to certain findings, 4-1BBζ-CART may have a longer half-life than CD28ζ-CART. However, the second was also observed to result in constitutive stimulation (activity in the absence of the antigen), even if the first may produce early depletion of CART. Because of this, CAR designs have improved to become more effective costimulatory structures.15
Third-generation CART
Several end domains of costimulatory signaling domains have been combined to create third-generation CARs. These structures are known to exist in CD3ζ-CD28-OX40 and CD3ζ-CD28-41BB. Given that 4-1BB end domains support the long-term survival of CARs and CD28 costimulatory domains are known to cause fast tumor eradication, the latter was very encouraging. In contrast to second-generation CART, no improved efficacy was attained, despite the fact that they have been utilized to treat several cancer types with favorable safety profiles, higher persistence, and proliferation.16,17
Fourth-generation CART
Fourth-generation CARs were based on second-generation designs since several costimulatory domains did not increase CART efficacy. In addition, the most recent generation is altered with a constitutive or inducible expression cassette that contains a transgenic protein, like a cytokine.
They are intended to transport the transgenic product to the intended tumor site and are known as T cell redirected for universal cytokine-mediated killing (TRUCK) CART. Usually, to accomplish this, these cells are engineered to carry a nuclear factor of activated T cell (NFAT)-responsive cassette, which contains a transgenic cytokine like IL-12. Therefore, when CD3ζ-containing CARs interact with their particular target, the transgene is expressed. Two transgenic cassettes must be transferred in order to create TRUCK CART in practice: one for the inducible cytokine and one for the CAR structure.18,19 Compared to second-generation CARs, the addition of a cytokine transgene significantly increased the effectiveness of CART therapies in preclinical animals. Additionally, the strategy proved effective in preventing systemic toxicity, which is one of the most frequent side effects of CART therapy.20
Fifth-generation CART
In an effort to improve efficacy, safety, proliferation, and durability, CART treatments have undergone significant change in recent years. It is still difficult to reduce the off-target and off-tumor toxicity of CART, nevertheless. Next-generation CART is being developed in this context. The new generation, which specialists have already dubbed the fifth generation, is distinct from the earlier ones due to the integration of an extra membrane receptor.21
Contact with the target antigen triggers the expression of the secondary transgene, which is followed by transcription and release into the extracellular fluid in TRUCKS, also known as fourth-generation CARs.22 In this method, the released signal reactivates the immune system to react to restimulation in addition to stimulating CART to stay active and produce memory T cells.23
CART that makes use of fifth-generation membrane receptors operates on a different concept. The inclusion of IL-2 receptors, which enables JAK/STAT pathway activation in an antigen-dependent manner, is the most promising of the various strategies presently being studied. Although other strategies have been tried, the discovery of switch receptors was one of the most fascinating advancements in the industry. There have been recent reports of successfully incorporating an ON switch that leads to activation or a drug-dependent OFF switch that leads to CAR depletion. Lenalidomide-gated CARs were created and evaluated using these guidelines. Despite having a little lower in vitro efficacy than previous generations of CARs, these cells were far easier to regulate, which improved their safety profile and expanded their therapeutic window.24
Targets of CART treatment for hematological tumors
A careful balance between safety and efficacy is required for the discovery of suitable tumor antigens, which is crucial for successful CART therapy. The following requirements should be fulfilled by preferred antigens: (I) show high and consistent surface expression on tumor cells, (II) continue to express themselves at different stages of the disease, (III) are important in the pathophysiology of the disease, (IV) shed little or nothing into the bloodstream, (V) are not impacted by certain treatment-induced pressures that could cause down-regulation or elimination, and (VI) do not express themselves on normal tissues.25-27
CD19. For the treatment of B-cell tumors, CD19 is thought to be the perfect target for CART immunotherapy. Normal tissues and cells do not express it; however, a range of B-cell malignant tumor cells and B-cell progenitor cells can. Grupp et al. used the second-generation CD19 CART (4-1BB/CD3ζ) to treat two children with B-cell ALL.28 About two months after treatment, one patient experienced a relapse with blast cells that no longer expressed CD19, whereas the other patient experienced total remission. Thirty patients with relapsed or refractory pediatric ALL experienced complete remission in a different clinical trial that used CAR to treat CD19 + B cell malignancy.7 B cell chronic lymphocytic leukemia (CLL) also expresses CD19, in addition to B cell ALL. Although several CARTs that are used to treat B-cell ALL are also evaluated to treat B-cell CLL, their therapeutic impact on B-cell CLL is not as strong. 29 In ten patients with chronic lymphoma/leukemia, Kochenderfer et al. found that autologous CD19-directed CART had anti-tumor effects.30 None of the patients experienced graft-versus-host disease (GVHD), and one patient experienced a total remission. After that, the group used CD19-specific CART (CD28/CD3ζ) to treat DLBCL patients. Four of the 15 patients experienced partial remission, while eight experienced total remission.31
CD20. Since the expression patterns of CD20 and CD19 are similar, adoptive immunotherapy for hematological tumors may benefit from CARTs that target CD20. Numerous research studies have currently employed anti-CD20 CART therapy as a therapeutic intervention.32 Researchers want to investigate the effectiveness of CD20-specific CAR autologous T cells in treating patients with relapsed indolent NHL and MCL (mast cell leukemia) in a phase I clinical trial. Using this method, the researcher showed adoptive T cell therapy's potential anticancer activity and safety. The effectiveness of second-generation CART therapy in DLBCL patients was uncertain. One research team addressed this by using anti-CD20 CART with 4-1BB for these patients, and the results of this innovative treatment were encouraging.33 According to the first report, three of the seven patients with refractory advanced CD20 + DLBCL who were enrolled experienced partial remission, and one experienced complete remission.
CD22. Apart from CD19 and CD20, clinical studies are also being conducted to examine CD22, which has been identified as a possible therapeutic target. For instance, individuals with B-ALL disorders who were not candidates for CD19-CART therapy have been observed to benefit from CD22-CART.34 Nonetheless, a large number of patients continue to relapse as a result of inadequate CART resistance or persistence. Therefore, several strategies were used to increase CD22-CART anti-cancer activity. A second-generation CAR with scFv binding a proximal CD22 epitope showed better antileukemic efficacy than previous binding domains.35
CD30. Clinical investigations are investigating CD30 as a distinctive marker of malignant Reed-Sternberg cells in HL.36 For patients with relapsed anaplastic large cell lymphoma (ALCL) who have not previously received it, Benuximab Vedotin, which binds an anti-CD30 antibody conjugated to monomethyl auristatin E (MMAE), would be the first choice. Additionally, it is the first FDA-approved medication for Hodgkin's lymphoma patients that was successfully created. Although it is far from definitive, numerous studies have shown that around half of patients with peripheral T cell lymphomas (PTCL) have high expression of CD30.37 Therefore, future treatment options for refractory or recurrent CD30-positive PTCL are made possible by CART therapy that targets CD30.
RORI. RORI, a receptor tyrosine kinase family member, is extensively expressed during embryonic development and in a variety of human cancers.38 Only adipocytes, the basal epithelial lining of the esophagus, the surface and foveolar epithelial cells of the stomach antral mucosa, and the duodenal mucosa exhibit RORI expression, which can be either positive or negative in normal tissues. Because of its tumor-selective expression, RORI may be a viable substitute for CART immunotherapy. A growing body of research indicates that ROR1 possesses intrinsic kinase activity, which mediates breast cancer bone metastases by interacting with the Hippo-YAP pathway.39 According to recent research by Lars et al, ROR1-CART have the ability to eradicate tumor cells that are stored in crypt formations.40
CD123. For hematolymphoid cancers, CD123 has become a new target. First, CD123 is widely overexpressed in hematolymphoid neoplasms, such as systemic macrocytosis, acute myeloid leukemia, acute lymphoblastic leukemia, blastic plasmacytoid dendritic cell neoplasm, and hairy cell leukemia.41 Second, a number of encouraging trial results demonstrated that CD123 modification holds great potential as a cancer immunotherapy tactic, particularly for CD123-targeting CART.42 As a result, efforts are being made to create CART treatments that specifically target hematological malignancies that express CD123. Brett M's group, for instance, created a CD123 CAR using a single-chain variable fragment that was obtained from a CD123 monoclonal antibody. Additionally, individuals with a range of hematological malignancies, such as plasmacytoid dendritic cell neoplasm and relapsed/refractory AML, have demonstrated noteworthy clinical success with these CD123 CART.43
CD33. Therapeutic targeting of CD33 has long been of interest. According to certain pre-clinical research, CD33-CARs significantly eradicate tumors and exhibit long-lasting, strong anti-tumor activity against AML cells.44 Wang et al showed in an early clinical trial that autologous T-cells expressing 38% anti-CD33 CAR would exhibit significant cytolytic activity against CD33+blasts. Targeting CD33, however, may provide challenges because myeloid cells and normal progenitor cells also express this protein. As a result, successful treatment is likely to be a major factor in delayed hematopoietic recovery and persistent cytopenia.45
Relapse and CART resistance
Although CART therapy has shown notable effectiveness in certain patients, it has drawbacks, including tumor recurrence and medication resistance in some people.46,47 Relapse rates can exceed 50% in DLBCL and range from 10% to 30% in B-ALL.48 Antigen escape, CART fatigue, and an immune-suppressive milieu are some of the factors that contribute to this (Fig. 1).
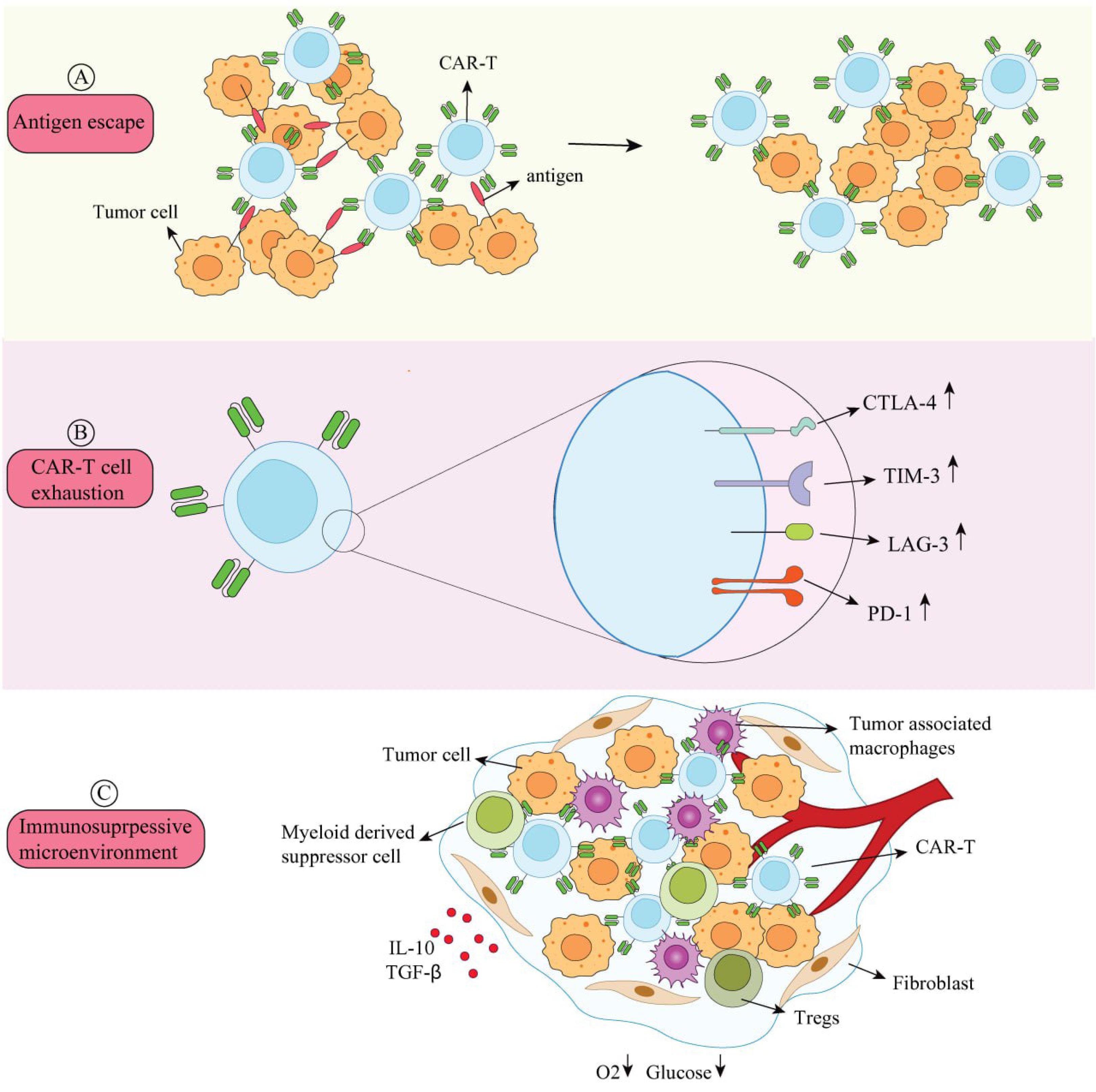
Fig. 1.
Mechanisms of CART resistance and relapse.(A) Antigen Escape: Antigens on the surface of tumor cells can evade detection and elimination. (B) CART Exhaustion: When CARTs express more inhibitory receptors (PD-1, TIM-3, LAG-3, and CTLA-4), their anti-tumor efficacy is diminished. (C) Immunosuppressive Microenvironment: The extracellular matrix, cytokines (TGF-β, IL-10), and immunosuppressive cells (TAMS, Tregs, and myeloid-derived suppressor cells) are all present in the tumor microenvironment. These elements restrict infiltration, impair CART activity, and suppress immune cells. CART activity is further decreased by tumor cells' consumption of glucose and oxygen, which results in hypoxia and nutrient deprivation.
.
Mechanisms of CART resistance and relapse.(A) Antigen Escape: Antigens on the surface of tumor cells can evade detection and elimination. (B) CART Exhaustion: When CARTs express more inhibitory receptors (PD-1, TIM-3, LAG-3, and CTLA-4), their anti-tumor efficacy is diminished. (C) Immunosuppressive Microenvironment: The extracellular matrix, cytokines (TGF-β, IL-10), and immunosuppressive cells (TAMS, Tregs, and myeloid-derived suppressor cells) are all present in the tumor microenvironment. These elements restrict infiltration, impair CART activity, and suppress immune cells. CART activity is further decreased by tumor cells' consumption of glucose and oxygen, which results in hypoxia and nutrient deprivation.
Antigen escape
Through strategies such as acquired mutations, selective splicing, and lineage shift, tumor cells avoid CART attacks during CART therapy, leading to altered or diminished surface antigen targets. Evasion is possible for common surface targets, including B-cell maturation antigen (BCMA), CD19, CD20, and CD22.49,50 Genetic alterations in CD19, which are present in the majority of resistant tumor cells, have been discovered by clinical analysis of relapsed samples. These mutations may result in protein truncation and subsequent loss of surface antigen.51 According to a number of studies, BCMA CART therapy-treated patients with relapsed MM show reduced BCMA surface expression on tumor cells.52 This process of antigen escape might complicate therapy and reduce treatment duration and patient survival. Therefore, in order to successfully address these issues, new treatment approaches are required.
CART exhaustion
Reduced CART cytotoxicity is the result of CART exhaustion, which is indicated by increased expression of inhibitory receptors on the surface of CART, including cytotoxic T lymphocyte antigen-4 (CTLA-4), lymphocyte activation gene-3 (LAG-3), programmed cell death protein-1 (PD-1), and T-cell immunoglobulin and mucin domain-containing protein-3 (TIM-3).53,54 While some individuals show positive responses when using CD19 CART to treat CLL, the majority of patients do not gain anything from CART therapy. T-cells in non-responsive patients upregulate pathways linked to effector differentiation, glycolysis, fatigue, and apoptosis, according to transcriptome sequencing.55 This exhaustion significantly affects CART functionality, suggesting that reducing exhaustion could enhance therapeutic efficacy. Research should aim to mitigate exhaustion to improve treatment outcomes. Reducing tiredness may improve therapeutic efficacy because it has a major impact on CART functioning. In order to enhance treatment results, research should focus on reducing fatigue.
Immunosuppressive microenvironment
The effects of CART are felt in the tumor microenvironment. An extracellular matrix, suppressor cytokines including TGF-β and IL-10, and a large number of immunosuppressive cells, including tumor-associated macrophages (TAMS), regulatory T-cells (Tregs), and myeloid suppressor cells, are also present.56 Through various ways, these components suppress immune-activated cells, impairing CART activity and preventing their invasion.57 Enhancing the effectiveness of CART treatment for hematologic malignancies requires research into how the immunosuppressive milieu affects CART therapy and the development of countermeasures. To overcome these obstacles, researchers are actively investigating novel strategies.58 Combining CART therapy with other medications is one such strategy that makes use of the advantages of different treatment techniques. This combined strategy can prolong the patients' survival by maintaining the effectiveness of CART and offsetting the drawbacks of CART monotherapy. This all-encompassing therapeutic approach increases hope and opportunities for cancer therapy by providing more comprehensive and efficient treatment options (Fig. 2).59,60
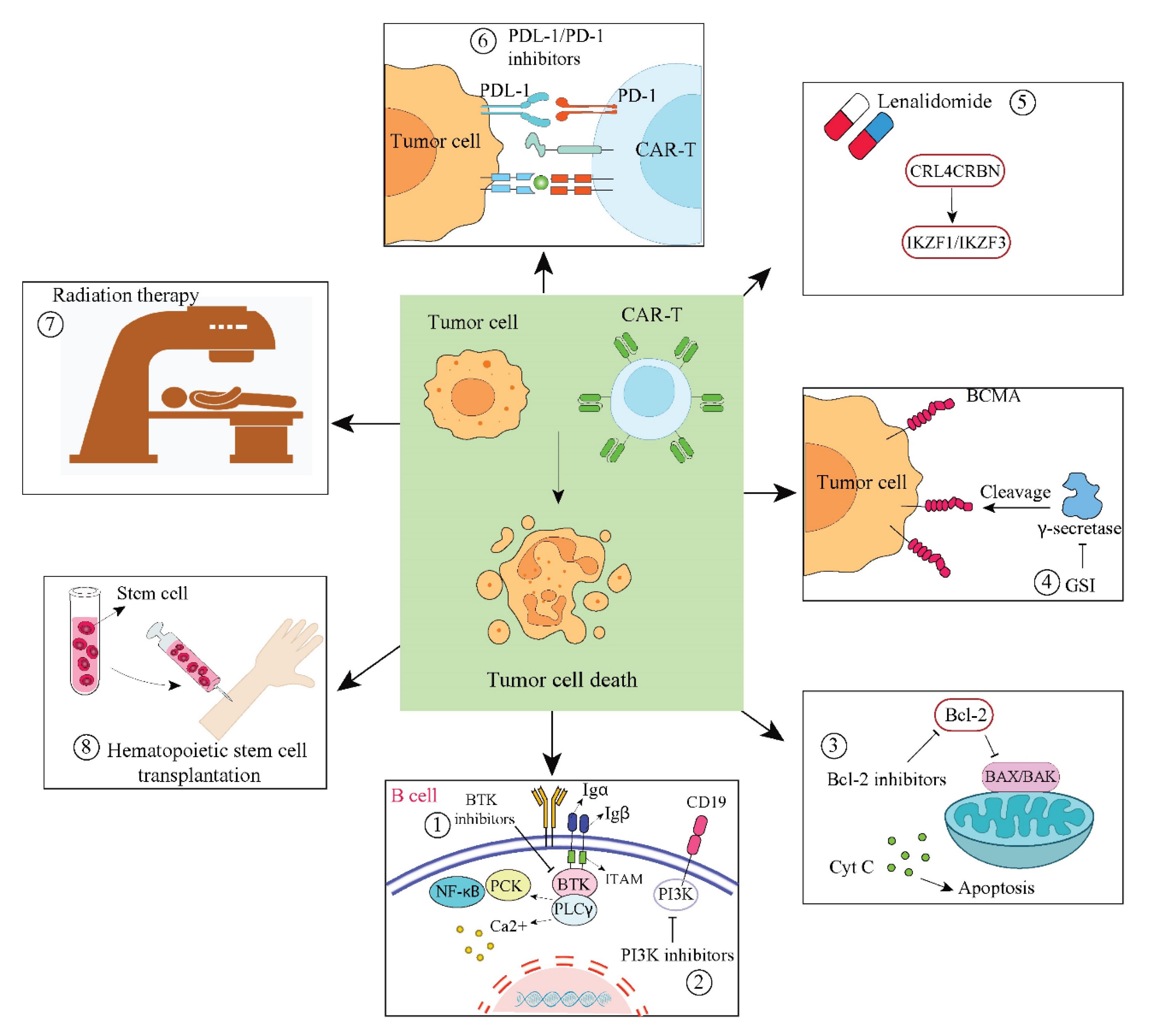
Fig. 2.
Combination Therapies with CARTs. (1) BTK inhibitors and CARTs work together to suppress BTK in the B-cell signaling pathway, which lowers the proliferation of malignant B cells. (2) By blocking the PI3K signaling pathway, the combination of CARTs with PI3K inhibitors prevents tumor cells from growing and surviving. (3) By blocking the BCL-2 protein, the combination of CARTs and BCL-2 inhibitors encourages tumor cell death. (4) By blocking γ-secretase, CARTs and GSI stop BCMA from being shed from tumor cell surfaces. (5) Lenalidomide and CARTs work together to attract the E3 ubiquitin ligase CRL4CRBN, which causes IKZF1 and IKZF3 to degrade. (6) By blocking the PD-1/PD-L1 signaling pathway, CARTs and PD-1/PD-L1 inhibitors work together to prevent tumor cells from evading the immune system. (7) The efficacy of CART therapy is increased when it is combined with radiation therapy, which destroys tumor cells directly. (8) The patient's immune system is restored when CARTs and hematopoietic stem cell transplantation are combined, supporting the long-lasting anti-tumor action of CARTs.61,62
.
Combination Therapies with CARTs. (1) BTK inhibitors and CARTs work together to suppress BTK in the B-cell signaling pathway, which lowers the proliferation of malignant B cells. (2) By blocking the PI3K signaling pathway, the combination of CARTs with PI3K inhibitors prevents tumor cells from growing and surviving. (3) By blocking the BCL-2 protein, the combination of CARTs and BCL-2 inhibitors encourages tumor cell death. (4) By blocking γ-secretase, CARTs and GSI stop BCMA from being shed from tumor cell surfaces. (5) Lenalidomide and CARTs work together to attract the E3 ubiquitin ligase CRL4CRBN, which causes IKZF1 and IKZF3 to degrade. (6) By blocking the PD-1/PD-L1 signaling pathway, CARTs and PD-1/PD-L1 inhibitors work together to prevent tumor cells from evading the immune system. (7) The efficacy of CART therapy is increased when it is combined with radiation therapy, which destroys tumor cells directly. (8) The patient's immune system is restored when CARTs and hematopoietic stem cell transplantation are combined, supporting the long-lasting anti-tumor action of CARTs.61,62
Anti-CD19 CART history
The most frequent cause of cancer in children is pediatric ALL.63 Cure rates above 80% are a result of scientific and clinical study. Unfortunately, there is a considerable mortality rate in this population, and some patients experience relapses and/or have refractory disease. With the addition of more salvage therapies, the risk of morbidity increases.64 Phase 1 and phase 2 CART studies were the result of focused immunotherapy research because of the possibility of dose-limiting toxicities and chemotherapy resistance. With a complete response (CR) rate of 93%, a phase 1-2 single-center anti-CD19 CART trial for children and young adults with relapsed or refractory B-cell ALL demonstrated encouraging outcomes. A phase 2 worldwide, multisite trial was created for this patient population in light of these findings. There were 25 locations across 11 nations in this experiment. From 2015 to 2017, patients were enrolled and given infusions. Median follow-up at 38.8 months showed that of 79 patients infused, there was an 82% overall remission rate, along with 3-year relapse-free survival (RFS) and overall survival rates of 52% and 63%, respectively. Long-term safety was shown to be favorable.64 The FDA approved Tisagenlecleucel, the first commercialized anti-CD19 CART therapy, in August 2017 for the treatment of pediatric relapsed or refractory B cell ALL in large part because to these pivotal phase 2 global, multisite study response rates and safety data. When compared to the phase 1-2 single center trial and the phase 2 global, multisite trial, the first real-world report of Tisagenlecleucel demonstrates comparable response rates and safety. The initial CR rate was 85.5%, and the 12-month duration of response was 60.9%, with a median follow-up of 13.4 months. Fifty-five percent of the patients experienced cytokine release syndrome (CRS), a systemic inflammatory reaction including increased cytokines in response to immune system activation. Twenty-seven percent of individuals had immune effector cell-associated neurotoxicity syndrome (ICANS).10. A phase 2, single-arm, multi-center trial in the Children's Oncology Group is investigating the use of Tisagenlecleucel up front for high-risk pediatric patients with positive minimal residual disease at the conclusion of consolidation.65
B cell hematologic malignancies
Recurrence of B cell ALL with CART treatment
Regretfully, there are two ways in which patients may relapse following anti-CD19-directed CART therapy. With CD19-positive B cell ALL, which still expresses the CD19 antigen, a patient may experience a relapse. This frequently occurs when CART exhibits transient persistence, which eliminates disease surveillance. CART fatigue or immune-mediated rejection may be linked to short-term persistence. The lack of CD19 expression on the tumour cell surface may result in CD19-negative relapses. Following anti-CD19 CART therapy, patient results for B-cell ALL relapse are not good. Newer CART trials have been developed to address these concerns as a result of research into strategies for B-cell ALL relapse after CART therapy.
Humanized CART
One indicator of CART persistence is B cell aplasia, an on-target/off-tumor toxicity that occurs after CART infusion. CART persistence is lost when B-cell aplasia is lost.66 At least 25% of patients undergoing CART infusions experience a loss of CART persistence. Twelve B cell recovery during the first six months indicates an early loss of CART persistence, which is associated with poor outcomes for these patients.66 A murine monoclonal antibody is the source of the extracellular antigen-binding domain seen in the majority of CARTs, which may trigger an antimurine immune response that results in CART rejection. To prevent this immune rejection and enhance long-term persistence, research has been conducted and is still being conducted on substituting a humanized component.67 One institution's investigation demonstrated that humanized CART can assist patients in achieving persistent remission. The backbone of Tisagenlecleucel served as the basis for this CAR. There were two cohorts in this study: CAR naïve and retreatment. Patients with non-response, early B cell recovery, or CD19 relapse who had previously received a murine CART product were included in the retreatment cohort. Overall response rate (ORR) for patients with B cell aplasia at day 28 was 64% in the retreatment cohort. RFS was 74% at month 12 and 58% at month 24. The CR rate was 98% at day 28, the RFS was 84% at month 12, and 74% at month 24 in the CAR naïve sample, which included patients who had never received a CART product.67 These findings suggest that this treatment holds promise.
Universal CART
A high number of circulating blasts, age at diagnosis, and intensive chemotherapy pretreatment are among the characteristics that make it very difficult for some patients to successfully collect and manufacture T-cells.68,69 According to a study, patients who have received extensive pretreatment may exhibit deficiencies in naïve T-cells, which may contribute to harvesting and manufacturing failure. Additionally, naïve and early memory T-cells in the collection product are correlated with good CART performance. Concerns have also been raised over the caliber of T-cells obtained from baby ALL patients.70 Cost, manufacturing time, T cell malfunction, and disease progression and death during the manufacturing process are further obstacles to autologous CART therapy.71 Successful product manufacturing is not always feasible for people with extremely aggressive illnesses.
Products made from T-cells obtained from healthy allogeneic donors are known as universal CART. "Off-the-shelf" CART are another name for these cells. For patients with highly aggressive diseases or those who have had unsuccessful T-cell collections, this is a promising treatment that may be used before an autologous CART product is available.72 These cells have the advantages of being less expensive, producing several doses from a single donor, not requiring chemotherapy, and being ready for use right away.73 Additionally, there are several drawbacks to commercially available CART products. Both GVHD and universal CART rejection are risks; as a result, these cells will not be as persistent as autologous CART. In order to prepare the patient's immune system to minimize rejection of the CART, alemtuzumab, an anti-CD52 immunotherapy, is frequently added to the lymphodepleting chemotherapy to intensify it. This immunosuppression increases the risk of infection. Viral reactivation may result from marrow suppression and protracted cytopenia.73
The feasibility of universal CART therapy was demonstrated by a universal CART trial that included patients ranging in age from 9 months to 62 years from 2016 to 2018. Eleven days was the median amount of time between trial consent and the onset of lymphodepletion.74 Seven children and fourteen adults with relapsed or refractory B cell ALL were involved in this experiment. Of the patients, 91% had CRS, 38% had ICANS, and 10% had GVHD. Some patients had viral infections, such as BK virus, adenovirus, human metapneumovirus, and CMV. Seventy-one percent of the responding patients underwent stem cell transplantation (SCT), and sixty-seven percent achieved CR 28 days after CART infusion.13 Results from a second phase 1 global CART trial were released recently. Participants in this research ranged in age from six months to eighteen years. Four of the six infused patients were in remission and underwent SCT. In the first several hours after CART injection, viral reactivation, ICANS, and controllable CRS were observed.75 Both trials demonstrate that universal CART therapy is safe, and practical, and has potential for individuals for whom autologous CART therapy is not an option, but further research is required to make meaningful judgements about efficacy.
Anti-CD22 CARTs
As was already established, CD19-negative illness can recur in individuals who experience a relapse following anti-CD19 CART therapy. Research indicates that within the first year of receiving anti-CD19 CART treatments, almost 50% of patients may experience a recurrence.76 These relapses are CD19 negative in about 40% of cases. These people require alternative therapies because their options for therapy are restricted. CD19-negative relapses have been considered the primary mechanism of resistance since the introduction of anti-CD19 immunotherapies. The leukemia cells typically keep their expression of CD22 even when they no longer exhibit the CD19 antigen.
The majority of B-cell malignancies exhibit the antigen CD22, which is only expressed by B cells in healthy tissue.50 Trials of anti-CD22 CARTs are now underway. Responses against leukemia have been observed, and the safety profile is comparable to that of anti-CD19 CARs.
Recently, the findings of a phase 1 anti-CD22 CART experiment were made public. 87.9% of the fifty-eight patients who received anti-CD19 CART were injected. 32.8% of patients experienced moderate neurotoxicity, while 86.2% of patients experienced CRS. The percentage of total remission was 70%. Relapse was eventually experienced by 75% of these patients. Patients who had previously received CD22 targeted therapy were shown to have lower MRD negative full remission rates and shorter remission durations.
Dual CART
Dual-targeting CART have become possible due to anti-CD22 CART trials that provide anti-leukemia responses for relapsed and refractory B cell ALL. Multi-agent chemotherapy is a key component of the initial treatment of ALL in order to prevent relapses caused by medication resistance.50 One of the main problems with CART treatment is CD19 escape, which can occur when single antigen-targeting drugs are used. In an effort to overcome CD19 antigen escape and enhance results, dual CARs that target the CD19 and CD22 antigens have been produced.77 There are four strategies to generate dual CARTs: tandem, bicistronic, co-transduction, and coadministration.78 Investigations are ongoing to find the best manufacturing and administration techniques.
There is no additional toxicity, according to dual CAR experiments that target both CD19 and CD22. Recently, the results of a dual CART experiment that targeted both CD19 and CD22 were made public. 194 patients received infusions, and 225 patients under the age of 20 were included in a phase 2 experiment. A 99% full response rate was observed. At 12 months, overall survival was 87.7%, and event-free survival (EFS) was 73.5%. Patients with B cell aplasia and those who went on to SCT had improved EFS, suggesting that CART persistence lasted longer than six months. ICANS affected 20.9% of individuals, while CRS affected 88%. There were three fatalities during CRS and/or ICANS. 78 The trial's findings demonstrated the effectiveness of this dual CART product, which assisted patients in achieving long-lasting remissions.
B cell non-Hodgkin lymphoma
Chemotherapy produces great results for the majority of pediatric patients with mature B-cell non-Hodgkin lymphoma(NHL). Although they are uncommon, recurrent illness has a poor prognosis and reduced chemosensitivity.79,80 One intriguing new treatment option for this patient group is CART therapy. Trials are currently being conducted to examine the safety and effectiveness of CART treatment for B cell NHL, which targets CD19, CD20, and CD22. In children with relapsed or refractory B-cell NHL, Tisagenlecleucel was found to be safe and effective in phase 2 international multicenter research. Although large B-cell lymphoma patients had a superior ORR (46%) than those with Burkitt lymphoma (20%), it was still encouraging. CRS happened in 70% of patients, while neurologic problems happened in 27% of individuals.
Non-B cell hematologic malignancies
It has proven more difficult to create effective CART treatments for non-B-cell hematological malignancies. Finding tumor-specific antigens to target and preventing severe, unacceptable on-target/off-tumor toxicities that kill healthy, normal cells have been some of the difficulties. Anti-T-cell therapy causes T-cell aplasia. Unacceptable long-term off-tumor toxicities include myeloid aplasia brought on by anti-AML CART and ALL CART.
T-cell ALL
The discovery of effective treatments for T-cell ALL has proven more challenging, despite the success of CART therapy for B-cell ALL. Ten to fifteen percent of ALL patients are T-cell ALL.81 With an overall survival rate of less than 10%, relapsed and refractory T-cell ALL has a poor prognosis and is infamously difficult to treat.82 CART fratricide (the self-destruction of CARTs), on-target/off-tumor toxicity of T-cell aplasia, which results in life-threatening immunodeficiency, and CART product contamination with T lymphoblasts are some of the special difficulties associated with CARTs for T-cell ALL.83
Autologous and universal CARTs are being used in early-stage experiments.84 There have been encouraging results from a recent phase 1 human universal anti-CD7 CART experiment. The issue of product contamination with leukemic cells is mitigated by using a universal CART from a healthy donor. They injected twenty patients. There were no toxicities that were limited by dosage. 90% of patients experienced a complete response (CR), the CARTs proliferated, and the therapy was shown to be safe.81
The findings encourage more research on CARTs for T-cell ALL that has relapsed and is refractory. It is too early in the treatment process to determine whether CART therapy alone will cure T-cell ALL without the need for SCT.
Acute myeloid leukemia
Chemotherapy resistance makes treating relapsed and refractory AML difficult. Alternative therapies are required for this patient population because SCT is the only possible curative therapy. 85 Finding a targetable antigen that does not result in intolerable toxicity is one of the same difficulties facing CART therapy for use in AML as it is in T ALL. Normal myeloid progenitors contain the majority of targetable antigens. Life-threatening infections and severe immunosuppression may result from the destruction of these progenitors.82,85 CD33, CLL-1, and CD123 are targets being investigated for relapsed and refractory AML.
The majority of AML cells express CD33, and the success of treating pediatric AML with Gemtuzumab, an anti-CD33 antibody drug combination, gives hope that anti-CD33 CART will work.86 The extent of myeloid toxicity, its reversibility, and its capacity for recovery are currently being investigated and determined. To better assess this, more experiments are needed.82 Research is being done to develop CARTs that reduce the degree and length of myelosuppression in AML patients who have relapsed or are refractory.85 Following CART therapy, the majority of trials recommend SCT because of the expected on-target/off-tumor effects. Suicide genes, safety switches that disable the CART, and genetic inactivation of certain cells are being investigated in other investigations.85,87 At the moment, the majority of AML CART trials seek to elicit remission and prepare the patient for SCT.
Conclusion
Over the past 11 years, pediatric oncology has seen significant advancements thanks to CART therapy. Patients with relapsed and refractory B-cell ALL were the first to get this revolutionary treatment. Many patients find this therapy to be promising and helpful, yet some still relapse. More trials are being developed for B cell ALL relapses following CART therapy since these patients need new treatments. Trials for further relapsed and refractory paediatric hematologic cancers have also been prompted by this outcome.
Review Highlights
What is the current knowledge?
What is new here?
-
In high-risk patients with refractory or multiple relapsing illnesses, CART treatment is still utilized as salvage therapy despite its great efficacy. To determine the best time to administer CART within the current therapy paradigm, clinical trials are still being conducted.
Competing Interests
The author declares that no conflict of interest.
Ethical Approval
Not applicable.
References
- Zhou Y, Li L, Yu Z, Gu X, Pan R, Li Q. Dermatophagoides pteronyssinus allergen Der p 22: cloning, expression, IgE-binding in asthmatic children, and immunogenicity. Pediatr Allergy Immunol 2022; 33:e13835. doi: 10.1111/pai.13835 [Crossref] [ Google Scholar]
- Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014; 123:2625-35. doi: 10.1182/blood-2013-11-492231 [Crossref] [ Google Scholar]
- Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015; 125:4017-23. doi: 10.1182/blood-2014-12-580068 [Crossref] [ Google Scholar]
- Levin A, Shah NN. Chimeric antigen receptor modified T cell therapy in B cell non-Hodgkin lymphomas. Am J Hematol 2019; 94:S18-s23. doi: 10.1002/ajh.25403 [Crossref] [ Google Scholar]
- Song EZ, Milone MC. Pharmacology of chimeric antigen receptor-modified T cells. Annu Rev Pharmacol Toxicol 2021; 61:805-29. doi: 10.1146/annurev-pharmtox-031720-102211 [Crossref] [ Google Scholar]
- Yan T, Zhu L, Chen J. Current advances and challenges in CAR T-cell therapy for solid tumors: tumor-associated antigens and the tumor microenvironment. Exp Hematol Oncol 2023; 12:14. doi: 10.1186/s40164-023-00373-7 [Crossref] [ Google Scholar]
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507-17. doi: 10.1056/NEJMoa1407222 [Crossref] [ Google Scholar]
- Lee JB, Chen B, Vasic D, Law AD, Zhang L. Cellular immunotherapy for acute myeloid leukemia: how specific should it be?. Blood Rev 2019; 35:18-31. doi: 10.1016/j.blre.2019.02.001 [Crossref] [ Google Scholar]
- Talleur AC, Myers R, Annesley C, Shalabi H. Chimeric antigen receptor T-cell therapy: current status and clinical outcomes in pediatric hematologic malignancies. Hematol Oncol Clin North Am 2022; 36:701-27. doi: 10.1016/j.hoc.2022.03.005 [Crossref] [ Google Scholar]
- Locke FL, Mahmoudjafari Z, Kebriaei P, Gardner RA, Frigault MJ, Frey NV, et al. Awakening from REMS: ASTCT 80/20 ongoing recommendations for safe use of chimeric antigen receptor T cells. Transplant Cell Ther 2025; 31: 349.e1-12. doi: 10.1016/j.jtct.2025.02.009.
- Malissen B, Bongrand P. Early T cell activation: integrating biochemical, structural, and biophysical cues. Annu Rev Immunol 2015; 33:539-61. doi: 10.1146/annurev-immunol-032414-112158 [Crossref] [ Google Scholar]
- Malissen B, Grégoire C, Malissen M, Roncagalli R. Integrative biology of T cell activation. Nat Immunol 2014; 15:790-7. doi: 10.1038/ni.2959 [Crossref] [ Google Scholar]
- Blache U, Popp G, Dünkel A, Koehl U, Fricke S. Potential solutions for manufacture of CAR T cells in cancer immunotherapy. Nat Commun 2022; 13:5225. doi: 10.1038/s41467-022-32866-0 [Crossref] [ Google Scholar]
- Zhang C, Wang L, Zhang Q, Shen J, Huang X, Wang M. Screening and characterization of the scFv for chimeric antigen receptor T cells targeting CEA-positive carcinoma. Front Immunol 2023; 14:1182409. doi: 10.3389/fimmu.2023.1182409 [Crossref] [ Google Scholar]
- Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer 2019; 120:26-37. doi: 10.1038/s41416-018-0325-1 [Crossref] [ Google Scholar]
- Hossian A, Hackett CS, Brentjens RJ, Rafiq S. Multipurposing CARs: same engine, different vehicles. Mol Ther 2022; 30:1381-95. doi: 10.1016/j.ymthe.2022.02.012 [Crossref] [ Google Scholar]
- Johnson PC, Abramson JS. Engineered T cells: CAR T cell therapy and beyond. Curr Oncol Rep 2022; 24:23-31. doi: 10.1007/s11912-021-01161-4 [Crossref] [ Google Scholar]
- Al-Haideri M, Tondok SB, Safa SH, Maleki AH, Rostami S, Jalil AT. CAR-T cell combination therapy: the next revolution in cancer treatment. Cancer Cell Int 2022; 22:365. doi: 10.1186/s12935-022-02778-6 [Crossref] [ Google Scholar]
- Huynh D, Winter P, Märkl F, Endres S, Kobold S. Beyond direct killing-novel cellular immunotherapeutic strategies to reshape the tumor microenvironment. Semin Immunopathol 2023; 45:215-27. doi: 10.1007/s00281-022-00962-4 [Crossref] [ Google Scholar]
- Liu Y, Adu-Berchie K, Brockman JM, Pezone M, Zhang DK, Zhou J. Cytokine conjugation to enhance T cell therapy. Proc Natl Acad Sci U S A 2023; 120:e2213222120. doi: 10.1073/pnas.2213222120 [Crossref] [ Google Scholar]
- Xie B, Li Z, Zhou J, Wang W. Current status and perspectives of dual-targeting chimeric antigen receptor T-cell therapy for the treatment of hematological malignancies. Cancers (Basel) 2022; 14:3230. doi: 10.3390/cancers14133230 [Crossref] [ Google Scholar]
- Li S, Ling S, Wang D, Wang X, Hao F, Yin L, et al. Modified lentiviral globin gene therapy for pediatric β0/β0 transfusion-dependent β-thalassemia: a single-center, single-arm pilot trial. Cell Stem Cell 2024; 31: 961-73.e8. doi: 10.1016/j.stem.2024.04.021.
- Balagopal S, Sasaki K, Kaur P, Nikolaidi M, Ishihara J. Emerging approaches for preventing cytokine release syndrome in CAR-T cell therapy. J Mater Chem B 2022; 10:7491-511. doi: 10.1039/d2tb00592a [Crossref] [ Google Scholar]
- Hamieh M, Mansilla-Soto J, Rivière I, Sadelain M. Programming CAR T cell tumor recognition: tuned antigen sensing and logic gating. Cancer Discov 2023; 13:829-43. doi: 10.1158/2159-8290.Cd-23-0101 [Crossref] [ Google Scholar]
- Abbott RC, Cross RS, Jenkins MR. Finding the keys to the CAR: identifying novel target antigens for T cell redirection immunotherapies. Int J Mol Sci 2020; 21:515. doi: 10.3390/ijms21020515 [Crossref] [ Google Scholar]
- Wang C, Wang J, Che S, Zhao H. CAR-T cell therapy for hematological malignancies: history, status and promise. Heliyon 2023; 9:e21776. doi: 10.1016/j.heliyon.2023.e21776 [Crossref] [ Google Scholar]
- Fang W, Sun W, Fang W, Zhao S, Wang C. Clinical features, treatment, and outcomes of patients with carfilzomib induced thrombotic microangiopathy. Int Immunopharmacol 2024. 134: 112178. doi: 10.1016/j.intimp.2024.112178.
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368:1509-18. doi: 10.1056/NEJMoa1215134 [Crossref] [ Google Scholar]
- Braendstrup P, Levine BL, Ruella M. The long road to the first FDA-approved gene therapy: chimeric antigen receptor T cells targeting CD19. Cytotherapy 2020; 22:57-69. doi: 10.1016/j.jcyt.2019.12.004 [Crossref] [ Google Scholar]
- Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013; 122:4129-39. doi: 10.1182/blood-2013-08-519413 [Crossref] [ Google Scholar]
- Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015; 33:540-9. doi: 10.1200/jco.2014.56.2025 [Crossref] [ Google Scholar]
- Ramos CA, Heslop HE, Brenner MK. CAR-T cell therapy for lymphoma. Annu Rev Med 2016; 67:165-83. doi: 10.1146/annurev-med-051914-021702 [Crossref] [ Google Scholar]
- Wang Y, Zhang WY, Han QW, Liu Y, Dai HR, Guo YL. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin Immunol 2014; 155:160-75. doi: 10.1016/j.clim.2014.10.002 [Crossref] [ Google Scholar]
- Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 2019; 33:2854-66. doi: 10.1038/s41375-019-0488-7 [Crossref] [ Google Scholar]
- Zanetti SR, Velasco-Hernandez T, Gutierrez-Agüera F, Díaz VM, Romecín PA, Roca-Ho H. A novel and efficient tandem CD19- and CD22-directed CAR for B cell ALL. Mol Ther 2022; 30:550-63. doi: 10.1016/j.ymthe.2021.08.033 [Crossref] [ Google Scholar]
- Moskowitz CH, Nademanee A, Masszi T, Agura E, Holowiecki J, Abidi MH. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin's lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015; 385:1853-62. doi: 10.1016/s0140-6736(15)60165-9 [Crossref] [ Google Scholar]
- Bossard C, Dobay MP, Parrens M, Lamant L, Missiaglia E, Haioun C. Immunohistochemistry as a valuable tool to assess CD30 expression in peripheral T-cell lymphomas: high correlation with mRNA levels. Blood 2014; 124:2983-6. doi: 10.1182/blood-2014-07-584953 [Crossref] [ Google Scholar]
- Chien HP, Ueng SH, Chen SC, Chang YS, Lin YC, Lo YF. Expression of ROR1 has prognostic significance in triple negative breast cancer. Virchows Arch 2016; 468:589-95. doi: 10.1007/s00428-016-1911-3 [Crossref] [ Google Scholar]
- Zhao Y, Zhang D, Guo Y, Lu B, Zhao ZJ, Xu X. Tyrosine kinase ROR1 as a target for anti-cancer therapies. Front Oncol 2021; 11:680834. doi: 10.3389/fonc.2021.680834 [Crossref] [ Google Scholar]
- Wallstabe L, Göttlich C, Nelke LC, Kühnemundt J, Schwarz T, Nerreter T. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 2019; 4:e126345. doi: 10.1172/jci.insight.126345 [Crossref] [ Google Scholar]
- Perriello VM, Gionfriddo I, Rossi R, Milano F, Mezzasoma F, Marra A. CD123 is consistently expressed on NPM1-mutated AML cells. Cancers (Basel) 2021; 13:496. doi: 10.3390/cancers13030496 [Crossref] [ Google Scholar]
- Jacoby E, Shahani SA, Shah NN. Updates on CAR T-cell therapy in B-cell malignancies. Immunol Rev 2019; 290:39-59. doi: 10.1111/imr.12774 [Crossref] [ Google Scholar]
- Budde L, Song JY, Kim Y, Blanchard S, Wagner J, Stein AS. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: a first-in-human clinical trial. Blood 2017; 130:811. doi: 10.1182/blood.V130.Suppl_1.811.811 [Crossref] [ Google Scholar]
- Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia 2014; 28:1596-605. doi: 10.1038/leu.2014.62 [Crossref] [ Google Scholar]
- Maakaron JE, Rogosheske J, Long M, Bachanova V, Mims AS. CD33-targeted therapies: beating the disease or beaten to death?. J Clin Pharmacol 2021; 61:7-17. doi: 10.1002/jcph.1730 [Crossref] [ Google Scholar]
- Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT. Three-year follow-up of KTE-X19 in patients with relapsed/refractory mantle cell lymphoma, including high-risk subgroups, in the ZUMA-2 study. J Clin Oncol 2023; 41:555-67. doi: 10.1200/jco.21.02370 [Crossref] [ Google Scholar]
- Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol 2022; 23:91-103. doi: 10.1016/s1470-2045(21)00591-x [Crossref] [ Google Scholar]
- Nastoupil LJ, Jain MD, Feng L, Spiegel JY, Ghobadi A, Lin Y. Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: results from the US lymphoma CAR T consortium. J Clin Oncol 2020; 38:3119-28. doi: 10.1200/jco.19.02104 [Crossref] [ Google Scholar]
- Aparicio-Pérez C, Carmona M, Benabdellah K, Herrera C. Failure of ALL recognition by CAR T cells: a review of CD 19-negative relapses after anti-CD 19 CAR-T treatment in B-ALL. Front Immunol 2023; 14:1165870. doi: 10.3389/fimmu.2023.1165870 [Crossref] [ Google Scholar]
- Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 2018; 24:20-8. doi: 10.1038/nm.4441 [Crossref] [ Google Scholar]
- Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med 2018; 24:1504-6. doi: 10.1038/s41591-018-0146-z [Crossref] [ Google Scholar]
- Lee H, Ahn S, Maity R, Leblay N, Ziccheddu B, Truger M. Mechanisms of antigen escape from BCMA- or GPRC5D-targeted immunotherapies in multiple myeloma. Nat Med 2023; 29:2295-306. doi: 10.1038/s41591-023-02491-5 [Crossref] [ Google Scholar]
- Aggarwal V, Workman CJ, Vignali DAA. LAG-3 as the third checkpoint inhibitor. Nat Immunol 2023; 24:1415-22. doi: 10.1038/s41590-023-01569-z [Crossref] [ Google Scholar]
- Agarwal S, Aznar MA, Rech AJ, Good CR, Kuramitsu S, Da T, et al. Deletion of the inhibitory co-receptor CTLA-4 enhances and invigorates chimeric antigen receptor T cells. Immunity 2023; 56: 2388-407.e9. doi: 10.1016/j.immuni.2023.09.001.
- Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018; 24:563-71. doi: 10.1038/s41591-018-0010-1 [Crossref] [ Google Scholar]
- Arner EN, Rathmell JC. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell 2023; 41:421-33. doi: 10.1016/j.ccell.2023.01.009 [Crossref] [ Google Scholar]
- Allen GM, Frankel NW, Reddy NR, Bhargava HK, Yoshida MA, Stark SR. Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science 2022; 378:eaba1624. doi: 10.1126/science.aba1624 [Crossref] [ Google Scholar]
- Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol 2020; 17:147-67. doi: 10.1038/s41571-019-0297-y [Crossref] [ Google Scholar]
- Zhou D, Zhu X, Xiao Y. CAR-T cell combination therapies in hematologic malignancies. Exp Hematol Oncol 2024; 13:69. doi: 10.1186/s40164-024-00536-0 [Crossref] [ Google Scholar]
- Yang Z, Liu X, Xu H, Teschendorff AE, Xu L, Li J. Integrative analysis of genomic and epigenomic regulation reveals miRNA mediated tumor heterogeneity and immune evasion in lower grade glioma. Commun Biol 2024; 7:824. doi: 10.1038/s42003-024-06488-9 [Crossref] [ Google Scholar]
- Zhou C, Kuang M, Tao Y, Wang J, Luo Y, Fu Y, et al. Nynrin preserves hematopoietic stem cell function by inhibiting the mitochondrial permeability transition pore opening. Cell Stem Cell 2024; 31: 1359-75.e8. doi: 10.1016/j.stem.2024.06.007.
- Li Z, Fan J, Xiao Y, Wang W, Zhen C, Pan J. Essential role of Dhx16-mediated ribosome assembly in maintenance of hematopoietic stem cells. Leukemia 2024; 38:2699-708. doi: 10.1038/s41375-024-02423-3 [Crossref] [ Google Scholar]
- Foster JB, Maude SL. New developments in immunotherapy for pediatric leukemia. Curr Opin Pediatr 2018; 30:25-9. doi: 10.1097/mop.0000000000000572 [Crossref] [ Google Scholar]
- Laetsch TW, Maude SL, Rives S, Hiramatsu H, Bittencourt H, Bader P. Three-year update of tisagenlecleucel in pediatric and young adult patients with relapsed/refractory acute lymphoblastic leukemia in the ELIANA trial. J Clin Oncol 2023; 41:1664-9. doi: 10.1200/jco.22.00642 [Crossref] [ Google Scholar]
- Schroeder BA, Jess J, Sankaran H, Shah NN. Clinical trials for chimeric antigen receptor T-cell therapy: lessons learned and future directions. Curr Opin Hematol 2022; 29:225-32. doi: 10.1097/moh.0000000000000723 [Crossref] [ Google Scholar]
- Pulsipher MA, Han X, Maude SL, Laetsch TW, Qayed M, Rives S. Next-generation sequencing of minimal residual disease for predicting relapse after tisagenlecleucel in children and young adults with acute lymphoblastic leukemia. Blood Cancer Discov 2022; 3:66-81. doi: 10.1158/2643-3230.Bcd-21-0095 [Crossref] [ Google Scholar]
- Myers RM, Li Y, Barz Leahy A, Barrett DM, Teachey DT, Callahan C. Humanized CD19-targeted chimeric antigen receptor (CAR) T cells in CAR-naive and CAR-exposed children and young adults with relapsed or refractory acute lymphoblastic leukemia. J Clin Oncol 2021; 39:3044-55. doi: 10.1200/jco.20.03458 [Crossref] [ Google Scholar]
- Sun DY, Hu YJ, Li X, Peng J, Dai ZJ, Wang S. Unlocking the full potential of memory T cells in adoptive T cell therapy for hematologic malignancies. Int Immunopharmacol 2025; 144:113392. doi: 10.1016/j.intimp.2024.113392 [Crossref] [ Google Scholar]
- Sun W, Jang MS, Zhan S, Liu C, Sheng L, Lee JH. Tumor-targeting and redox-responsive photo-cross-linked nanogel derived from multifunctional hyaluronic acid-lipoic acid conjugates for enhanced in vivo protein delivery. Int J Biol Macromol 2025; 314:144444. doi: 10.1016/j.ijbiomac.2025.144444 [Crossref] [ Google Scholar]
- Das RK, Vernau L, Grupp SA, Barrett DM. Naïve T-cell deficits at diagnosis and after chemotherapy impair cell therapy potential in pediatric cancers. Cancer Discov 2019; 9:492-9. doi: 10.1158/2159-8290.Cd-18-1314 [Crossref] [ Google Scholar]
- Graham C, Jozwik A, Pepper A, Benjamin R. Allogeneic CAR-T cells: more than ease of access?. Cells 2018; 7:155. doi: 10.3390/cells7100155 [Crossref] [ Google Scholar]
- Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov 2020; 19:185-99. doi: 10.1038/s41573-019-0051-2 [Crossref] [ Google Scholar]
- Benjamin R, Graham C, Yallop D, Jozwik A, Mirci-Danicar OC, Lucchini G. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. Lancet 2020; 396:1885-94. doi: 10.1016/s0140-6736(20)32334-5 [Crossref] [ Google Scholar]
- Liu B, Du H, Zhang J, Jiang J, Zhang X, He F. Developing a new sepsis screening tool based on lymphocyte count, international normalized ratio and procalcitonin (LIP score). Sci Rep 2022; 12:20002. doi: 10.1038/s41598-022-16744-9 [Crossref] [ Google Scholar]
- Ottaviano G, Georgiadis C, Gkazi SA, Syed F, Zhan H, Etuk A. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci Transl Med 2022; 14:eabq3010. doi: 10.1126/scitranslmed.abq3010 [Crossref] [ Google Scholar]
- Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL. CD4/CD8 T-cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a phase I anti-CD22 CAR T-cell trial. J Clin Oncol 2020; 38:1938-50. doi: 10.1200/jco.19.03279 [Crossref] [ Google Scholar]
- Wang T, Tang Y, Cai J, Wan X, Hu S, Lu X. Coadministration of CD19-and CD22-directed chimeric antigen receptor T-cell therapy in childhood B-cell acute lymphoblastic leukemia: a single-arm, multicenter, phase II trial. J Clin Oncol 2023; 41:1670-83. doi: 10.1200/jco.22.01214 [Crossref] [ Google Scholar]
- Shah NN, Maatman T, Hari P, Johnson B. Multi targeted CAR-T cell therapies for B-cell malignancies. Front Oncol 2019; 9:146. doi: 10.3389/fonc.2019.00146 [Crossref] [ Google Scholar]
- Pearson AD, Rossig C, Mackall C, Shah NN, Baruchel A, Reaman G. Paediatric strategy forum for medicinal product development of chimeric antigen receptor T-cells in children and adolescents with cancer: ACCELERATE in collaboration with the European Medicines Agency with participation of the Food and Drug Administration. Eur J Cancer 2022; 160:112-33. doi: 10.1016/j.ejca.2021.10.016 [Crossref] [ Google Scholar]
- Liu Y, Deng B, Hu B, Zhang W, Zhu Q, Liu Y. Sequential different B-cell antigen-targeted CAR T-cell therapy for pediatric refractory/relapsed Burkitt lymphoma. Blood Adv 2022; 6:717-30. doi: 10.1182/bloodadvances.2021004557 [Crossref] [ Google Scholar]
- Pan J, Tan Y, Wang G, Deng B, Ling Z, Song W. Donor-derived CD7 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia: first-in-human, phase I trial. J Clin Oncol 2021; 39:3340-51. doi: 10.1200/jco.21.00389 [Crossref] [ Google Scholar]
- Lamble AJ, Gardner R. CAR T cells for other pediatric non-B-cell hematologic malignancies. Hematology Am Soc Hematol Educ Program 2020; 2020:494-500. doi: 10.1182/hematology.2020000134 [Crossref] [ Google Scholar]
- Teachey DT, Hunger SP. Anti-CD7 CAR T cells for T-ALL: impressive early-stage efficacy. Nat Rev Clin Oncol 2021; 18:677-8. doi: 10.1038/s41571-021-00556-3 [Crossref] [ Google Scholar]
- Callahan C, Haas L, Smith L. CAR-T cells for pediatric malignancies: past, present, future and nursing implications. Asia Pac J Oncol Nurs 2023; 10:100281. doi: 10.1016/j.apjon.2023.100281 [Crossref] [ Google Scholar]
- Harris K, LaBelle JL, Bishop MR. Current status of CAR T cell therapy for leukemias. Curr Treat Options Oncol 2021; 22:62. doi: 10.1007/s11864-021-00859-8 [Crossref] [ Google Scholar]
- Fathi E, Farahzadi R, Sheervalilou R, Sanaat Z, Vietor I. A general view of CD33 + leukemic stem cells and CAR-T cells as interesting targets in acute myeloblatsic leukemia therapy. Blood Res 2020; 55:10-6. doi: 10.5045/br.2020.55.1.10 [Crossref] [ Google Scholar]
- Epperly R, Gottschalk S, Velasquez MP. Harnessing T cells to target pediatric acute myeloid leukemia: CARs, BiTEs, and beyond. Children (Basel) 2020; 7:14. doi: 10.3390/children7020014 [Crossref] [ Google Scholar]