Bioimpacts. 16:32638.
doi: 10.34172/bi.32638
Review
Integrin-linked kinase (ILK) in hematologic malignancies: Bridging molecular mechanisms to therapeutic innovation
Omer Qutaiba B. Allela Conceptualization, Investigation, Supervision, Writing – original draft, Writing – review & editing, 1, * 
Abdulkareem Shareef Writing – original draft, 2
Ashishkumar Kyada Writing – original draft, 3
H. Malathi Writing – original draft, 4
Laxmidhar Maharana Writing – original draft, 5
Dinesh Puri Writing – original draft, 6, 7
Harshit Gupta Writing – original draft, 8
Djamila Polatova Writing – original draft, 9
Hayder Naji Sameer Writing – original draft, 10
Ahmed Yaseen Writing – original draft, 11
Zainab H. Athab Writing – original draft, 12
Mohaned Adil Writing – original draft, 13
Author information:
1College of Pharmacy, Alnoor University, Mosul, Iraq
2Ahl al bayt University, Kerbala, Iraq
3Marwadi University Research Center, Department of Pharmaceutical Sciences, Faculty of Health Sciences, Marwadi University, Rajkot, Gujarat, India
4Department of Biotechnology and Genetics, School of Sciences, JAIN (Deemed to be University), Bangalore, Karnataka, India
5Department of Pharmaceutical Sciences, Siksha 'O' Anusandhan (Deemed to be University), Bhubaneswar, Odisha-751030, India
6Department of Pharmacy, Graphic Era Hill University, Dehradun, India
7Centre for Promotion of Research, Graphic Era Deemed to be University, Dehradun, Uttarakhand-248002, India
8Centre for Research Impact & Outcome, Chitkara University Institute of Engineering and Technology, Chitkara University, Rajpura, 140401, Punjab, India
9Scientific-Practical Medical Center for Pediatric Oncology, Hematology and Immunology, Tashkent, Uzbekistan
10Collage of Pharmacy, National University of Science and Technology, Dhi Qar, 64001, Iraq
11Gilgamesh Ahliya University, Baghdad, Iraq
12Department of Pharmacy, Al-Zahrawi University College, Karbala, Iraq
13Pharmacy College, Al-Farahidi University, Baghdad, Iraq
Abstract
Therapy resistance remains a formidable challenge in hematologic malignancies despite significant advances in targeted therapies. This comprehensive review examines integrin-linked kinase (ILK) as a critical molecular hub at the nexus of cell adhesion, signal transduction, and therapy resistance across leukemias, lymphomas, and multiple myeloma. Unlike in solid tumors, where ILK primarily drives invasion and metastasis, in hematologic malignancies it uniquely mediates microenvironmental protection and therapy resistance through distinct signaling networks. ILK functions as a central mediator connecting microenvironmental signals to intracellular survival pathways, with expression levels 5-20-fold higher in malignant cells compared to normal counterparts. Through systematic analysis of structural properties, expression patterns, downstream signaling, and microenvironmental interactions, we present compelling evidence for ILK as a promising therapeutic target capable of overcoming resistance mechanisms. Current data demonstrate that ILK inhibition simultaneously disrupts multiple survival pathways, sensitizes resistant cells to established therapies, and selectively targets therapy-resistant leukemic stem cells while sparing normal progenitors. This review provides a comprehensive framework for translating ILK-targeted approaches into innovative therapeutic strategies with significant potential to improve outcomes in treatment-refractory hematologic malignancies.
Graphical Abstract
Keywords: Integrin-linked kinase, Hematologic malignancies, Therapy resistance, Tumor microenvironment, Leukemic stem cells
Copyright and License Information
© 2026 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
This study did not receive any specific grants from funding agencies in the public, commercial, or not-for-profit sectors.
Introduction
Hematologic malignancies represent a diverse group of cancers affecting blood, bone marrow, and lymphatic tissues.1,2 Despite remarkable advances in treatment approaches—ranging from targeted kinase inhibitors to revolutionary immunotherapies—therapy resistance inevitably emerges in many patients, leading to relapse and disease progression.3,4 The underlying mechanisms of this resistance are multifactorial, involving both intrinsic cellular adaptations and protective signals from the tumor microenvironment.5,6 Recent evidence indicates that malignant hematopoietic cells exploit specialized niches within the bone marrow and lymphoid tissues, where they receive critical pro-survival signals that enable them to withstand therapeutic interventions.7,8
At the molecular level, integrin-linked kinase (ILK) has emerged as a pivotal orchestrator of these survival signals.9,10 ILK serves as a multifunctional protein that links integrin receptors to downstream pathways driving cancer cell survival, proliferation, and migration.9,11 While ILK's role in solid tumor progression has been extensively documented, its specific contributions to hematologic malignancies have only recently been appreciated.9,10 Unlike solid tumors, where ILK primarily drives invasion and metastasis through epithelial-mesenchymal transition, in hematologic malignancies it uniquely mediates microenvironmental protection and therapy resistance through distinct signaling networks.9,10
The clinical significance of ILK in hematologic malignancies is underscored by its overexpression across various blood cancers and its association with poor prognosis.9,10 ILK has been implicated in multiple resistance mechanisms, including those against Bruton's tyrosine kinase (BTK) inhibitors in chronic lymphocytic leukemia (CLL),12,13 conventional chemotherapies in acute myeloid leukemia (AML), and even cutting-edge immunotherapies like chimeric antigen receptor (CAR) T-cells.14-16
Understanding ILK's role in hematologic malignancies requires examination of its molecular architecture, interaction partners, and the signaling networks it regulates. By dissecting these aspects, we can identify potential vulnerabilities and develop strategies to target ILK-mediated resistance mechanisms.
Molecular architecture and functional organization of ILK
Integrin-linked kinase possesses a sophisticated molecular structure that facilitates its diverse cellular functions.17,18 ILK comprises three distinct domains, each serving specific roles in protein-protein interactions and signal transduction.19,20 The N-terminal region contains four ankyrin (ANK) repeats that mediate interactions with several binding partners, most notably PINCH.20,21 This interaction creates a stable complex that is necessary for the assembly of focal adhesions and cell survival signaling pathways (Table 1).
Table 1.
Structural and Functional Overview of ILK
|
Domain
|
Key features
|
Functional roles
|
Implications in hematologic malignancies
|
| Ankyrin repeat domain (ARD) |
Contains four ankyrin repeats |
Mediates interactions with PINCH & parvin |
Stabilizes focal adhesions, regulates cytoskeletal organization20,21 |
| Pleckstrin homology (PH)-like domain |
Binds PIP3, regulated by PTEN |
Facilitates membrane recruitment and activation |
Enables response to microenvironmental signals in bone marrow niches17,22 |
| Pseudokinase domain |
Lacks catalytic activity but binds AKT & GSK-3β |
Regulates PI3K/AKT, β-catenin signaling |
Drives survival, proliferation, and therapy resistance23,24 |
| C-terminal domain |
Integrin-binding, membrane localization |
Transduces ECM signals to intracellular pathways |
Enhances cell migration, invasion, and leukemic stem cell survival23,25 |
Between the ankyrin repeats and the C-terminal domain lies ILK's pleckstrin homology (PH)-like domain.17,22 This central region serves a crucial function by binding to phosphatidylinositol-3,4,5-triphosphate (PIP3), a lipid product generated by phosphoinositide 3-kinase (PI3K).17 This interaction facilitates ILK's recruitment to the plasma membrane, enabling its activation and subsequent downstream signaling.22
Perhaps most intriguing is ILK's C-terminal pseudokinase domain, which resembles a serine/threonine kinase domain but lacks key catalytic residues typically required for enzymatic activity.23,24 Despite its classification as a pseudokinase, this domain serves vital functions in protein-protein interactions. It binds to the cytoplasmic tails of β-integrins, creating a physical link between integrins and the actin cytoskeleton that influences cell shape, adhesion, and motility.
ILK's connection with several adaptor proteins that alter its activity and link it to various signaling networks further broadens its functioning (Fig. 1). ILK is connected to receptor tyrosine kinases (RTKs) and PI3K by NCK2 and insulin receptor substrate (IRS) proteins, creating a signaling axis that encourages cell survival and proliferation.26-28 Conversely, ILK-associated phosphatase (ILKAP) binds to the ankyrin repeats of ILK and functions as a negative regulator by dephosphorylating downstream targets.29,30
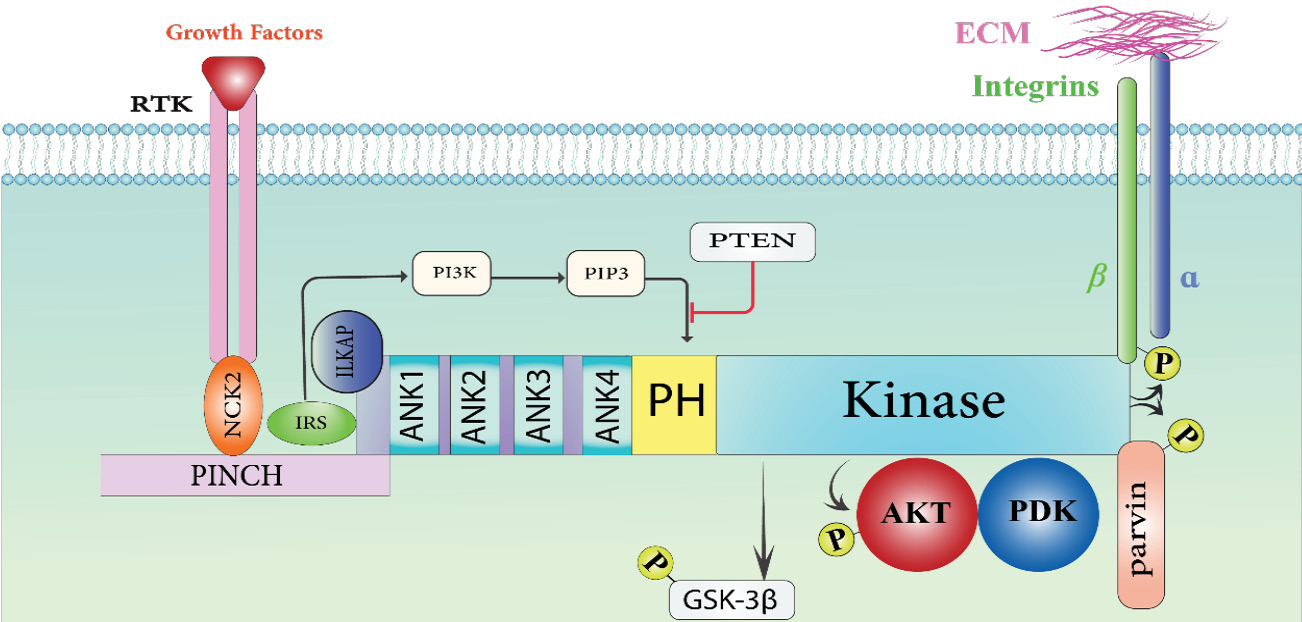
Fig. 1.
The interaction between Integrin-linked kinase (ILK) occurs within cells. ILK contains three different domains: the Ser-Thr kinase domain at the C-terminal, four ankyrin (ANK) repeats at the N-terminal, and the pleckstrin homology (PH) motif between these two domains. The five LIM domains that make up Cys-His-rich protein (PINCH) interact with ILK's Ank1. ILK is coupled to PI3K and the tyrosine kinase receptor (RTK) by the adaptor proteins NCK2 and IRS. ILK's kinase activity is inhibited by the interaction between Ank1 and the associated protein (ILKAP). Through Phosphatidylinositol-3, 4, 5-triphosphate (PIP3), PI3K attaches itself to the PH domain of ILK. By dephosphorylating PIP3 and ILKAP, PTEN functions as a negative regulator of ILK activity. The cytoplasmic domain of beta integrin is one of the intracellular proteins that the kinase domain of ILK interacts with. ILK can phosphorylate PKB, GSK-3, and Parvin, and it can also interact with signaling proteins like Akt/PKB, PDK-1, and GSK-3.
.
The interaction between Integrin-linked kinase (ILK) occurs within cells. ILK contains three different domains: the Ser-Thr kinase domain at the C-terminal, four ankyrin (ANK) repeats at the N-terminal, and the pleckstrin homology (PH) motif between these two domains. The five LIM domains that make up Cys-His-rich protein (PINCH) interact with ILK's Ank1. ILK is coupled to PI3K and the tyrosine kinase receptor (RTK) by the adaptor proteins NCK2 and IRS. ILK's kinase activity is inhibited by the interaction between Ank1 and the associated protein (ILKAP). Through Phosphatidylinositol-3, 4, 5-triphosphate (PIP3), PI3K attaches itself to the PH domain of ILK. By dephosphorylating PIP3 and ILKAP, PTEN functions as a negative regulator of ILK activity. The cytoplasmic domain of beta integrin is one of the intracellular proteins that the kinase domain of ILK interacts with. ILK can phosphorylate PKB, GSK-3, and Parvin, and it can also interact with signaling proteins like Akt/PKB, PDK-1, and GSK-3.
Recent structural studies using crystallography and cryo-electron microscopy have provided deeper insights into the precise molecular interactions within the ILK pseudokinase domain31 These studies reveal specific binding pockets and interface regions that can be targeted by small molecule inhibitors with improved specificity, facilitating the rational design of next-generation ILK inhibitors.31,32
ILK expression/role in hematologic malignancies
The ILK expression is significantly elevated across diverse hematologic malignancies compared to normal hematopoietic cells, suggesting its critical involvement in disease pathogenesis.9 ILK expression is significantly elevated in multiple myeloma (MM), acute myeloid leukemia (AML), chronic myeloid leukemia (CML), acute lymphoblastic leukemia (ALL), and lymphoid malignancies, such as diffuse large B-cell lymphoma (DLBCL), T-cell acute lymphoblastic leukemia (T-ALL), and CLL, according to quantitative analyses.9,10
Flow cytometry and immunofluorescence studies reveal that ILK levels are approximately 5-20-fold higher in malignant cells compared to normal bone marrow mononuclear cells.23,24 In CML, ILK overexpression correlates with disease aggressiveness and resistance to tyrosine kinase inhibitors (TKIs).9,10 K562 and KU812F cell lines show approximately 8.5-fold and 9-fold increases in ILK expression, respectively. Notably, imatinib-resistant CML cell lines (LAMA84R) exhibit even higher ILK levels (12-fold increase) compared to their imatinib-sensitive counterparts (LAMA84S, 7-fold increase), directly implicating ILK in TKI resistance mechanisms.33,34
Similarly, in AML, high ILK expression drives enhanced survival pathways and confers resistance to apoptosis-inducing chemotherapy agents. AML cell lines MOLM14 and NOMO-1 show 7.5-fold and 9.5-fold increases in ILK expression, respectively. This overexpression is particularly pronounced in FLT3-mutated AML, where ILK serves as a critical downstream effector of FLT3 signaling.35-39
Dysregulation of ILK in specific hematologic malignancies provides critical insights into its role in disease pathophysiology and therapy resistance. In CLL, ILK plays a crucial role in mediating resistance to Bruton's tyrosine kinase (BTK) inhibitors such as ibrutinib.40,41 While these targeted agents achieve high initial response rates in CLL and mantle cell lymphoma, complete remissions are rare, and residual disease eventually leads to relapse.42 Beyond acquired BTK mutations, resistance mechanisms involve the extrinsic protection conferred by the tumor microenvironment, with ILK serving as a critical mediator of this protection.12,43-45
In AML, ILK is essential for maximal Akt activation downstream of FLT3, a receptor tyrosine kinase frequently mutated in this disease.46,47 Inhibiting ILK reduces stromal-induced Akt signaling and impairs leukemic growth in vivo, suggesting that ILK helps AML cells resist tyrosine kinase inhibitors.48,49 Furthermore, ILK activation in AML triggers an IL-6/STAT3/NF-κB feedback loop, elevating IL-6 and IL-1β levels and fostering an immunosuppressive microenvironment.50,51
Multiple myeloma likewise leverages ILK-mediated adhesion mechanisms to evade therapy.52,53 Cell adhesion-mediated drug resistance is associated with high VLA-4 expression on myeloma cells, and integrin interaction in the bone marrow niche activates the PI3K/Akt, MAPK, and NF-κB pathways, which shield myeloma cells from chemotherapy.54,55 So, ILK serves as a critical mediator of these adhesion-induced survival signals, making it an attractive therapeutic target in this disease.56,57
On the other hand, in CLL, ILK overexpression enhances leukemia–stromal adhesion via integrin β1 signaling, resulting in Akt activation and GSK-3β inhibition, which together sustain NF-κB and β-catenin–dependent transcription of anti-apoptotic genes (BCL2, MCL1).58 By enhancing carcinogenic feedback loops that sustain proliferation and cell cycle progression through CCND1 and β-catenin activation, ILK functions downstream of aberrant Notch1 and PI3K/Akt pathways in T-ALL. By suppressing Akt signaling and encouraging caspase-mediated apoptosis, blocking ILK highlights how crucial it is for preserving T-ALL cell survival.59 ILK stimulates the development of the integrin–IKK–ILK complex, which promotes cytokine production, survival, and resistance to apoptosis, and corresponds with constitutive NF-κB and STAT3 activity in DLBCL, particularly the activated B-cell (ABC) subtype.60
ILK downstream pathways orchestrating oncogenic signaling networks
ILK serves as a central hub in an intricate network of signaling pathways that collectively promote survival, proliferation, and therapy resistance in hematologic malignancies (Fig. 2).22,61 This section provides an in-depth analysis of these downstream pathways, highlighting how ILK coordinates their activation and cross-talk to maintain malignant cell survival.62,63
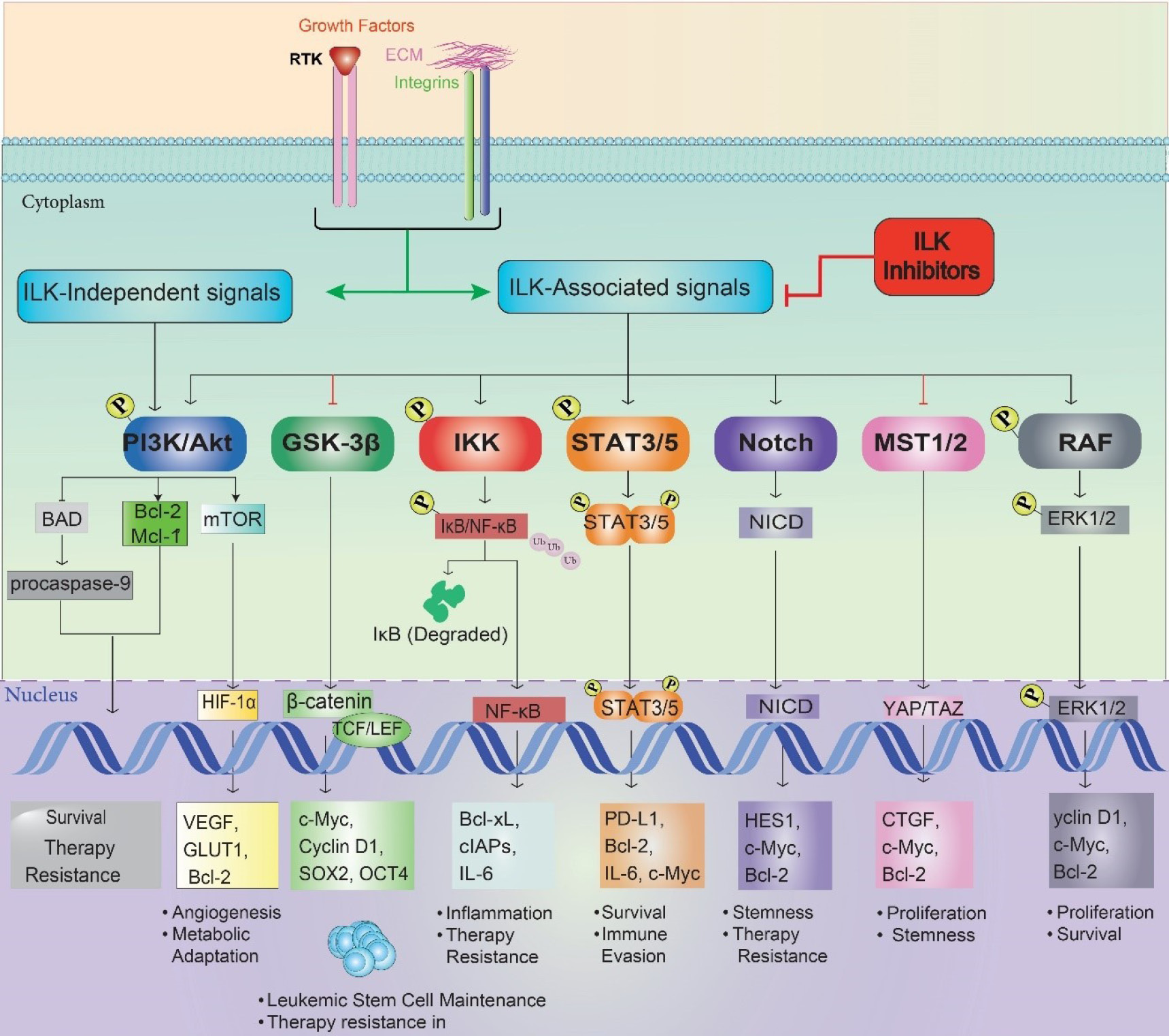
Fig. 2.
Cross-talk between ILK-driven signaling pathways in leukemic cells. This network diagram illustrates the interconnected signaling pathways downstream of ILK, emphasizing cross-talk interactions that drive oncogenesis in AML and CML. ILK (center, gray) orchestrates signaling in the cytoplasm (e.g., PI3K/Akt, RAF, MST1/2) and leads to the nuclear translocation of transcription factors in the nucleus (e.g., ERK1/2, β-catenin, NF-κB, STAT3/5, HIF-1α, NICD, YAP/TAZ). Pathways are color-coded: PI3K/Akt (blue), Wnt/β-catenin (green), NF-κB (red), STAT (orange), MST1/2 (Hippo) (yellow), RAF/MAPK/ERK (pink), Notch (teal), and Hypoxia/HIF-1α (purple). Dashed gray lines indicate cross-talk, such as Akt enhancing RAF activity, β-catenin synergizing with YAP/TAZ for stemness, and NF-κB collaborating with HIF-1α for VEGF expression, highlighting the complexity of ILK-driven oncogenic networks.
.
Cross-talk between ILK-driven signaling pathways in leukemic cells. This network diagram illustrates the interconnected signaling pathways downstream of ILK, emphasizing cross-talk interactions that drive oncogenesis in AML and CML. ILK (center, gray) orchestrates signaling in the cytoplasm (e.g., PI3K/Akt, RAF, MST1/2) and leads to the nuclear translocation of transcription factors in the nucleus (e.g., ERK1/2, β-catenin, NF-κB, STAT3/5, HIF-1α, NICD, YAP/TAZ). Pathways are color-coded: PI3K/Akt (blue), Wnt/β-catenin (green), NF-κB (red), STAT (orange), MST1/2 (Hippo) (yellow), RAF/MAPK/ERK (pink), Notch (teal), and Hypoxia/HIF-1α (purple). Dashed gray lines indicate cross-talk, such as Akt enhancing RAF activity, β-catenin synergizing with YAP/TAZ for stemness, and NF-κB collaborating with HIF-1α for VEGF expression, highlighting the complexity of ILK-driven oncogenic networks.
PI3K/Akt pathway: the master regulator of cell survival
The PI3K/Akt pathway represents one of the most critical signaling cascades regulated by ILK in hematologic malignancies.64 There is ongoing discussion on the exact mechanism by which ILK stimulates Akt phosphorylation. More recent research suggests that ILK largely acts as a molecular scaffold that moves Akt closer to its actual kinases, including mTORC2 or DNA-PK, despite prior research suggesting direct phosphorylation by ILK.65 Research by McDonald et al. demonstrated that ILK directly interacts with Rictor, a defining component of mTORC2, facilitating the phosphorylation of Akt at Ser473.66 Activated Akt phosphorylates and inactivates pro-apoptotic proteins including BAD and procaspase-9, while simultaneously promoting the expression of anti-apoptotic proteins such as Bcl-2 and Mcl-1.67,68 These changes dramatically raise the apoptotic threshold, making malignant cells resistant to a wide range of therapies.69,70 In AML, for instance, ILK-mediated Akt activation contributes to resistance against traditional chemotherapeutics like cytarabine and anthracyclines, while in CML it undermines the efficacy of TKIs such as imatinib.69,71 Beyond its direct effects on apoptotic machinery, Akt activation by ILK also promotes mTOR signaling, driving protein synthesis and metabolic reprogramming that further enhance malignant cell survival.72-74 This Akt-mTOR axis is particularly relevant in multiple myeloma, where it supports the excessive protein synthesis required by malignant plasma cells and contributes to proteasome inhibitor resistance.75-77
Wnt/β-catenin pathway: sustaining leukemic stem cells
ILK profoundly influences the Wnt/β-catenin pathway, which plays a crucial role in maintaining leukemic stem cells (LSCs) and promoting therapy resistance.78,79 The primary mechanism involves ILK-mediated inhibition of glycogen synthase kinase-3β (GSK-3β) through phosphorylation at Ser9.80,81 Inhibiting GSK-3β allows β-catenin to evade phosphorylation-dependent degradation, build up in the cytoplasm, and go into the nucleus, where it triggers the transcription of genes linked to survival, self-renewal, and stemness.82,83
This ILK-driven stabilization of β-catenin is particularly significant in the context of leukemic stem cells, which rely heavily on Wnt signaling for maintenance and self-renewal.84 By enhancing β-catenin activity, ILK preserves the LSC population even in the presence of therapies that effectively eliminate bulk leukemic cells.85 This mechanism helps explain the phenomenon of minimal residual disease and subsequent relapse observed in many patients with AML and CML.86-88
NF-κB signaling: inflammation and therapy resistance
ILK also plays a central role in activating NF-κB signaling, a pathway strongly associated with inflammation, survival, and therapy resistance in hematologic malignancies.63,89 This pathway is particularly relevant in B-cell malignancies such as CLL and multiple myeloma, where constitutive NF-κB activation drives disease progression and therapy resistance.90 In CLL, for instance, ILK-mediated NF-κB activation contributes to resistance against BTK inhibitors like ibrutinib, even in the absence of mutations in the drug's target.91
STAT signaling: a critical node in cytokine response
Beyond the three major pathways described above, ILK also regulates STAT (Signal Transducer and Activator of Transcription) signaling, particularly STAT3 and STAT5, which are critical mediators of cytokine responses in hematologic malignancies.92 The ILK-STAT3 axis is particularly relevant in AML and certain lymphomas, where constitutive STAT activation drives disease progression.48 Tabe et al demonstrated that coculture of leukemic cells with bone marrow stromal cells activates multiple signaling pathways, with ILK/STAT3 being a critical component.48 When ILK is inhibited, STAT3 phosphorylation is reduced by approximately 60%, leading to decreased expression of STAT3 target genes, including MCL1, survivin, and cyclin D1.46
Notch signaling: maintaining stemness and differentiation block
In normal hematopoiesis, Notch signaling regulates the balance between self-renewal and differentiation of stem and progenitor cells.93 In malignancies such as T-ALL and certain myeloid neoplasms, aberrant Notch activation contributes to differentiation arrest and maintenance of a stem-like phenotype.94,95 ILK modulates Notch signaling through regulation of Notch1 and its downstream effectors Hes1 and Hey1.46,96 Research by Tabe et al. revealed that ILK blockade significantly decreased Notch1 and Hes1 expression in leukemic cells cocultured with bone marrow stromal cells.46,96 This cross-talk is particularly evident in T-ALL, where ILK activation enhances Notch signaling, contributing to maintenance of leukemic stem cells and therapy resistance.97,98 By integrating Notch signaling with other pathways such as PI3K/Akt and Wnt/β-catenin, ILK creates a robust signaling network that sustains the malignant phenotype even under therapeutic pressure.99,100
Hippo pathway: controlling proliferation and organ size
The Hippo pathway, a conserved signaling cascade that regulates organ size and tissue homeostasis, represents another important downstream target of ILK in hematologic malignancies.101 ILK influences this pathway by inhibiting MST1/2 kinases, which are the upstream regulators of the Hippo pathway.102,103 This inhibition leads to reduced phosphorylation of YAP/TAZ, allowing them to accumulate in the nucleus and activate target genes involved in proliferation and survival.104,105 In certain leukemias and lymphomas, dysregulation of the Hippo pathway contributes to uncontrolled proliferation and resistance to therapy, with ILK serving as a critical modulator of this process.102,105,106 Targeting this ILK-Hippo axis could provide a novel approach to overcome therapy resistance, particularly in malignancies that show hyperactivation of YAP/TAZ signaling.107,108
MEK/ERK pathway: driving proliferation independently of Ras
In multiple myeloma and certain leukemias, ILK activates the MEK/ERK pathway independently of Ras mutations, contributing to cell proliferation and drug resistance.109,110 The ability of ILK to stimulate MEK/ERK signaling provides malignant cells with an alternative mechanism to maintain proliferative signals even when targeted therapies inhibit upstream components of the pathway.111,112 This is particularly relevant in the context of resistance to targeted therapies such as FLT3 inhibitors in AML or BCR-ABL inhibitors in CML, where activation of MEK/ERK through ILK can bypass the blockade of the primary oncogenic driver and sustain malignant cell proliferation.113,114 blockade of ILK signaling suppressed ERK1/2 activation in leukemic cells cocultured with bone marrow stromal cells, indicating that ILK is an important upstream regulator of the MEK/ERK pathway in the context of the bone marrow microenvironment.96,115 The ILK downstream pathways orchestrating oncogenic signaling networks has been summarized in Table 2.
Table 2.
Summary of ILK downstream pathways in hematologic malignancies
|
Pathway
|
Key mechanism of ILK regulation
|
Primary molecular effects
|
Biological consequences
|
Representative malignancies / resistance contexts
|
| PI3K/Akt |
ILK facilitates Akt phosphorylation at Ser473 via interaction with mTORC2 (Rictor) or DNA-PK. |
Activates Akt → Inhibits BAD, procaspase-9; Upregulates Bcl-2, Mcl-1; Activates mTOR. |
Increased survival, anti-apoptosis, metabolic reprogramming, therapy resistance. |
AML (resistance to cytarabine, anthracyclines), CML (imatinib resistance), Multiple myeloma (proteasome inhibitor resistance). |
| Wnt/β-catenin |
ILK phosphorylates and inhibits GSK-3β (Ser9), preventing β-catenin degradation. |
Stabilizes β-catenin → Nuclear translocation → Activation of stemness and survival genes (Cyclin D1, c-Myc, SOX2, OCT4, survivin). |
Maintains leukemic stem cells (LSCs), promotes minimal residual disease and relapse. |
AML, CML (stemness preservation, relapse origin). |
| NF-κB |
ILK promotes IKK activation → IκB degradation → NF-κB nuclear translocation. |
Transcription of Bcl-2, Bcl-xL, cIAPs; Cytokine induction (IL-6, TNF-α, IL-1β). |
Inflammation, anti-apoptosis, therapy resistance, microenvironmental support. |
CLL (resistance to ibrutinib), multiple myeloma. |
| STAT (STAT3/STAT5) |
ILK interacts with JAKs to enhance STAT3 Y705 phosphorylation and nuclear activity. |
Induces transcription of MCL1, survivin, cyclin D1, PD-L1. |
Promotes survival, proliferation, and immune evasion. |
AML, lymphomas (constitutive STAT signaling, PD-L1-mediated resistance). |
| Notch |
ILK upregulates Notch1 and downstream effectors Hes1 and Hey1. |
Sustains self-renewal, inhibits differentiation. |
Maintains stem-like phenotype, supports therapy resistance. |
T-ALL, myeloid neoplasms. |
| Hippo (YAP/TAZ) |
ILK inhibits MST1/2 kinases, reducing YAP/TAZ phosphorylation. |
Increases nuclear YAP/TAZ → Activation of proliferation and survival genes. |
Enhances proliferation, invasion, drug resistance. |
Leukemias, lymphomas (YAP/TAZ hyperactivation). |
| MEK/ERK |
ILK activates MEK/ERK via RAF interaction or PI3K/Akt cross-talk, independent of Ras. |
Stimulates ERK1/2 activation → Upregulates proliferation-related genes. |
Promotes proliferation, sustains growth under therapy pressure. |
AML (FLT3 inhibitor resistance), CML (BCR-ABL inhibitor resistance), multiple myeloma. |
Potential interplay between ILK signaling and extracellular vesicles in hematologic malignancies
The relationship between the ILK and extracellular vesicles (EVs) is fascinating and has a big impact on how hematologic cancers behave. As messengers, EVs—tiny membrane-bound packets released by cells—transport proteins, RNA, and other substances between cancer cells and the milieu around them.116 ILK-mediated pathways such as PI3K/Akt and NF-κB can be activated in recipient cells by integrins and associated signaling proteins found in EVs produced from malignant cells in hematologic malignancies. This activation increases the survival of cancer cells, fosters treatment resistance, and enables malignant cells to take advantage of their surroundings.117 ILK also affects the cytoskeletal configurations necessary for EV biogenesis and release, which means it can control the amount and caliber of EVs generated by tumor cells, thus strengthening these feedback loops. EVs derived from acute myeloid leukemia and chronic myeloid leukemia cells, for instance, activate anti-apoptotic proteins like BCL-xl and start autocrine signaling that promotes growth and resistance to chemotherapy.118 Additionally, EVs have the ability to rewire bone marrow niche stromal cells to take on a tumor-supporting phenotype by altering the extracellular matrix and secreting cytokines—processes that are closely related to ILK-driven integrin signaling. Therefore, there is a reciprocal relationship: EVs sustain ILK signaling in stromal cells and cancer cells, whereas ILK regulates EV generation and function. Leukemia progression, immunological evasion, and treatment resistance are all accelerated by this mutual interaction.119 Targeting ILK may disrupt these EV-mediated processes to reduce microenvironmental protection and overcome drug resistance in hematologic malignancies. Both ILK and EVs have the potential to be biomarkers and therapeutic targets, bringing new approaches to better treatment results for patients with blood malignancies, because they are detectable and accessible in patient samples. Further investigation of this interaction will enhance comprehension and improve therapeutic approaches.120
Epigenetic regulation and hypoxia response
ILK also exerts profound influences on epigenetic mechanisms and cellular responses to hypoxia, two processes that significantly impact therapy resistance in hematologic malignancies.121,122 Through regulation of histone deacetylases (HDACs) and DNA methyltransferases (DNMTs), ILK alters chromatin structure and gene expression patterns, contributing to the malignant phenotype and therapy resistance.123-125
The relationship between ILK and hypoxia response is particularly noteworthy. Under hypoxic conditions common in the bone marrow microenvironment, increased expression of hypoxia-inducible factor 1α (HIF-1α) enhances ILK expression and activation.126,127 Reciprocally, ILK stabilizes HIF-1α through inhibition of its proteasomal degradation pathway, specifically by reducing PHD activity and preventing VHL-mediated ubiquitination.128,129 This creates a positive feedback loop that amplifies hypoxia-mediated signals and promotes adaptation to low oxygen conditions through upregulation of genes involved in glycolysis, angiogenesis, and cell survival.130,131
ILK modulates VEGF expression by promoting HIF-1α protein expression through PKB/AKT and mTOR pathways, hence enhancing VEGF-induced angiogenesis.132,133 Moreover, mesenchymal stem cells co-cultured with myeloma cells in a hypoxic environment contribute to angiogenesis via the HIF-2α-ILK pathway.131,134 In this situation, HIF-2α enhances ILK, hence augmenting mesenchymal stem cell-mediated angiogenesis in multiple myeloma.131,135
The ILK-HIF axis is particularly significant in leukemic stem cells located in hypoxic bone marrow niches, as it facilitates metabolic adaptations and confers therapeutic resistance.136,137 ILK's capacity to modulate epigenetic processes and hypoxic responses affords malignant cells various protective layers against therapeutic obstacles, underscoring the intricacy of ILK-mediated resistance mechanisms.61,138
ILK in the tumor microenvironment and immune evasion
In addition to its intracellular signaling roles, ILK is essential in facilitating connections between malignant cells and the tumor microenvironment.139 The bone marrow niche, characterized by its intricate network of stromal cells, extracellular matrix constituents, and soluble molecules, offers a protective environment for hematologic malignancies.140 ILK functions as a vital intermediary in these protective relationships, converting adhesion signals into intracellular responses that enhance treatment resistance.141
Leukemic cells activate ILK and set in motion many downstream pathways that improve survival and resistance when they attach to stromal cells in the bone marrow or components of the extracellular matrix like fibronectin.48 Cell adhesion-mediated drug resistance (CAM-DR) is a big problem when it comes to treating blood cancers, especially AML and multiple myeloma.142,143
More recent developments in bone marrow microenvironment modeling have shed light on the function of ILK in this setting.144,145 Anatomical and cellular details of the native bone marrow niche, including stromal cells, osteoblasts, and vascular elements, were faithfully reproduced in the three-dimensional organoid model created by Baryawno and colleagues.146-148 Although it is usually difficult to sustain primary cells from various hematologic malignancies ex vivo, these organoids have effectively facilitated engraftment and survival of these cells.149,150 The significance of the three-dimensional microenvironment was highlighted by these investigations, which demonstrated that multiple myeloma cells lost vitality quickly in typical liquid culture settings but maintained > 90% viability after 12 days in bone marrow organoids.151,152 There is substantial evidence for a therapeutic window in a physiologically relevant environment, as these organoid models showed variable sensitivity patterns between malignant cells and normal hematopoietic progenitors when treated with ILK inhibitors.153,154
Not only does ILK affect conventional medication resistance, but it also affects immunotherapy failures. B-cell acute lymphoblastic leukemia and diffuse large B-cell lymphoma patients who undergo CAR T-cell therapy often experience remarkable remissions; yet, recurrence is common because tumor cells continue to elude immune clearance.155 By enhancing integrin-mediated adhesion and activating survival pathways, ILK strengthens this immunological privilege and makes tumor cells less vulnerable to T-cell cytotoxicity.17,156
Additional roles for ILK in immune checkpoint inhibitor resistance have been uncovered in recent research.157 In models of leukemia and lymphoma, it has been demonstrated that tumor cell ILK activation upregulates PD-L1 expression, which in turn contributes to immunological evasion and T cell exhaustion.158,159 When used in conjunction with immune checkpoint inhibitors such as anti-PD-1 antibodies, ILK inhibition improves T cell activation and tumor regression.160,161
Therapeutic targeting of ILK in hematologic malignancies
In vitro studies
In vitro effectiveness of various small-molecule ILK inhibitors against hematologic malignancies is encouraging.61 The three most researched inhibitors—Compound 22 (Cpd22), QLT0267, and OSU-T315—display different patterns of potency, selectivity, and mode of action.162-164
The MTT cell viability experiments reveal that Cpd22 is quite effective against chronic myeloid leukemia (CML) cell lines, with IC50 values varying between 235 and 500 Nm.165 Even at dosages that cause severe cell death in leukemic cells, this inhibitor shows remarkable selectivity, maintaining normal cell viability.49 It is worth mentioning that Cpd22 has the ability to circumvent existing resistance mechanisms. Its effectiveness is particularly demonstrated against imatinib-resistant CML cells (LAMA84R, IC50 = 300 nM).165 In AML cell lines, QLT0267 shows good activity at dosages up to 3000 nM (3 μM), however it is more concentration-dependent than Cpd22.166 Regardless, at 10 μM, normal bone marrow mononuclear cells retain 90% viability, demonstrating that QLT0267 retains selectivity for malignant cells.167
In the case of SUP-B15 (ALL) cells, OSU-T315 has strong activity, while in the case of MOLM14 (AML) cells, it demonstrates IC50 values of around 380 ± 40 nM.168 Unlike other ILK inhibitors, OSU-T315 causes cells to die in two distinct ways: via apoptosis and autophagy. This is quite intriguing.49 The phosphorylation of Akt at Ser473 (80% reduction) and GSK-3β at Ser9 (70% reduction)—essential nodes in the PI3K/Akt and Wnt/β-catenin signaling cascades—is markedly reduced after 72 hours of treatment with 400 nM Cpd22.164,169 Connected to these molecular occurrences are functional consequences, such as stem cell dysfunction, enhanced cell death, and cell cycle halt.49,162 Molecular effects of inhibiting ILK include halting cell cycle progression, inducing cell death, interfering with β-catenin signaling, blocking NF-κB signaling, and preventing STAT3 activation.47,48,170
The combination of Cpd22 with imatinib demonstrates strong synergy (Combination Index = 0.45) in CML cell lines, significantly enhancing apoptosis induction compared to either agent alone.171,172 Even more striking is the synergy observed between QLT0267 and dasatinib in imatinib-resistant CML cells (CI = 0.32), suggesting that this combination strategy may be particularly effective in the setting of established TKI resistance.173,174 The mechanistic basis for these synergistic interactions involves complementary pathway inhibition.22,61 While targeted therapies like TKIs primarily inhibit specific oncogenic drivers (e.g., BCR-ABL), ILK inhibitors simultaneously suppress alternative survival pathways such as PI3K/Akt and Wnt/β-catenin, preventing compensatory activation that often leads to resistance.162,169 Additionally, ILK inhibition disrupts adhesion-mediated drug resistance by interfering with integrin signaling and microenvironmental protection.48,175
In vivo (pre-clinical) efficacy and translation to clinical studies
There is strong evidence from animal model research that ILK inhibitors work in vivo. In CML xenograft models, treatment with Cpd22 (10 mg/kg) significantly reduced tumor volume (around 70%) and increased survival time.176 In AML xenograft models, QLT0267 (15 mg/kg) also improved overall survival and reduced tumor volume by 65%.177 The combination of Cpd22 with imatinib reduced tumor volume by 85% in TKI-resistant rats, demonstrating that combination treatments are even more effective in vivo.178
Promising preclinical data have led to preparations for first-in-human clinical studies. Currently in late preclinical research with Phase I trials expected to start in 2025–2026, the most advanced contender is a modified form of Cpd22 with these enhanced pharmacokinetic characteristics.179,180 With an eye toward safety, tolerability, and first efficacy, these initial studies will center on patients with relapsed/refractory CML and AML who have failed conventional therapy.181
Early safety evaluations in animal models are promising, despite the fact that pharmacokinetic factors and other toxicities pose obstacles to clinical translation.169 No notable toxicities were seen in complete blood counts, liver enzymes, or renal function tests at doses that achieved powerful anti-tumor effects, suggesting that ILK inhibitors were usually well-tolerated at therapeutic doses.182
Particularly important is new research by UBC and BC Cancer (2023) showing that ILK inhibition especially targets dormant cancer stem cells that normal TKI treatment cannot sufficiently eradicate on its own.183 Given most treatments that preferentially target actively dividing cells, these dormant cancer stem cells provide a significant obstacle in treating hematologic malignancies.184 While preserving healthy stem cells, ILK inhibitors combined with conventional treatments efficiently sensitize these drug-resistant cancer stem cells, thus addressing a major unmet demand in the treatment of hematologic malignancies.185 Overall, therapeutic targeting of ILK in hematologic malignancies has been summarized in Table 3.
Table 3.
Therapeutic targeting of ILK in hematologic malignancies
|
Section
|
Category
|
Key Findings / Details
|
| In vitro studies |
Lead ILK Inhibitors |
Compound 22 (Cpd22), QLT0267, OSU-T315 |
| Potency (IC50 Values) |
-Cpd22:235–500 nM in CML; 300 nM in imatinib-resistant CML (LAMA84R) -QLT0267: Up to 3 μM in AML -OSU-T315: ~380 ± 40 nM in MOLM14 (AML); strong activity in SUP-B15 (ALL) |
| Selectivity |
Cpd22 and QLT0267 maintain normal cell viability ( ≥ 90%) at effective doses. |
| Unique Features |
-Cpd22: Active against TKI-resistant CML cells -OSU-T315: Induces apoptosis and autophagy simultaneously |
| Molecular Mechanisms of Action |
- Inhibits ILK-mediated phosphorylation of Akt (Ser473, −80%) and GSK-3β (Ser9, −70%) - Disrupts PI3K/Akt, Wnt/β-catenin, NF-κB, and STAT3 signaling - Induces apoptosis, cell cycle arrest, and loss of leukemia stemness |
| Combination Synergy (In Vitro) |
-Cpd22 + Imatinib:CI = 0.45, enhanced apoptosis in CML -QLT0267 + Dasatinib:CI = 0.32, synergy in imatinib-resistant CML - Mechanistic basis: dual inhibition of oncogenic and compensatory survival pathways, plus disruption of adhesion-mediated resistance. |
| Preclinical in vivo models |
Efficacy in Animal Models |
-Cpd22 (10 mg/kg): Reduced CML xenograft tumor volume by 70%, improved survival. -QLT0267 (15 mg/kg): Reduced AML xenograft tumor volume by 65%, extended survival. -Cpd22 + Imatinib: Decreased tumor volume by 85% in TKI-resistant rodent models. |
| Stem Cell Impact |
ILK inhibition sensitizes dormant leukemia stem cells resistant to TKIs, while sparing normal hematopoietic stem cells (UBC & BC Cancer 2023). |
| Safety Profile (Preclinical) |
No significant hematologic, hepatic, or renal toxicities observed at efficacious doses. |
| Clinical setting |
Translation and Future Directions |
- Modified Cpd22 analog advancing toward Phase I clinical trials (2025–2026) for relapsed/refractory CML and AML.
- Early preclinical safety supports good tolerability and favorable pharmacokinetics. |
While the preclinical evidence supporting ILK inhibition in hematologic malignancies is robust, successful clinical translation demands a strategic framework that integrates biomarker-guided patient selection, rational combination regimens, vigilant safety monitoring, and alignment with emerging therapeutic modalities. Recent advances now position ILK not only as a mechanistic linchpin of microenvironment-mediated resistance but also as a tractable node for precision intervention. The modified analog of Compound 22 (Cpd22)—optimized for enhanced bone marrow penetration, metabolic stability, and pharmacokinetic profile—is advancing toward first-in-human Phase I trials focusing on patients with relapsed/refractory AML and CML who have failed standard therapies, including TKIs.181 Primary endpoints will assess safety, maximum tolerated dose, and preliminary efficacy signals such as reduction in minimal residual disease and leukemic stem cell depletion. Given the heterogeneity of ILK dependency across hematologic subtypes, predictive biomarkers are essential to enrich for likely responders. Emerging data support a composite biomarker strategy that includes ILK overexpression ( ≥ 5-fold vs. normal hematopoietic cells by qRT-PCR or IHC), phospho-Akt (Ser473) and phospho-GSK-3β (Ser9) as functional readouts of pathway activation, integrin β1/VLA-4 surface expression (particularly relevant in AML and multiple myeloma), and stromal adhesion signatures derived from bone marrow biopsies or circulating extracellular vesicles. These markers are being retrospectively validated and will be prospectively integrated into upcoming trials.79
Although ILK is ubiquitously expressed, malignant cells exhibit heightened dependency on ILK for survival and niche adhesion, creating a therapeutic window. However, chronic ILK inhibition may impair normal hematopoietic stem cell retention in the bone marrow niche, potentially leading to cytopenias or delayed hematopoietic recovery. To mitigate this, intermittent dosing schedules and short-course combination regimens are under evaluation. Additionally, bone marrow–targeted delivery systems—such as nanoparticles functionalized with CXCR4 or CD44 ligands—are being developed to maximize tumor exposure while minimizing systemic toxicity. ILK inhibition holds particular promise in combinatorial approaches that disrupt both oncogenic drivers and microenvironment-mediated resistance: in CML, ILK inhibitors synergize with TKIs (e.g., imatinib, dasatinib) by overcoming stroma-induced survival signals; in CLL, combining ILK inhibitors with BTK inhibitors (e.g., ibrutinib) may prevent microenvironment-driven NF-κB reactivation; in AML, ILK blockade sensitizes leukemic stem cells to venetoclax or chemotherapy by suppressing β-catenin and MCL-1; in multiple myeloma, ILK inhibition disrupts VLA-4–mediated adhesion and reverses proteasome inhibitor resistance; and with immunotherapies, ILK inhibition downregulates PD-L1 and enhances CAR T-cell cytotoxicity, supporting trials combining ILK inhibitors with anti–PD-1 or CD19-directed CAR T-cell products.16 These combinations are prioritized based on mechanistic synergy, non-overlapping toxicity, and unmet need in resistant/refractory settings. Collectively, the convergence of biomarker science, novel drug delivery, and rational polytherapy is transforming ILK from a compelling biological target into a viable clinical strategy. By simultaneously dismantling intrinsic survival pathways and extrinsic microenvironmental protection, ILK-targeted therapy offers a promising avenue to eradicate persistent disease reservoirs and improve long-term outcomes in high-risk hematologic malignancies.
Study limitations and challenges
There are a number of important obstacles that must be carefully considered when implementing ILK as a therapeutic target in clinical settings. The variable and context-dependent function of ILK in various hematologic malignancies is a major cause for worry. ILK inhibition is consistently effective against myeloid tumors, whereas its effects on lymphoid cancers are wildly inconsistent and contradictory.186 Interestingly, ILK overexpression causes some B-cell lymphomas to develop less rather than more, indicating that ILK might act as a context-dependent tumor suppressor in particular situations. This basic biological difference suggests that ILK-targeted treatments won't work for everyone and that a malignancy-specific—and possibly even genetic subtype-specific—therapeutic approach is needed.120
Additionally, a crucial unresolved issue with immediate therapeutic relevance is the long-term effects of ILK inhibition on normal hematopoiesis. Strong, long-term ILK inhibition may result in hematologic toxicities that are not entirely represented in short-term preclinical models, such as cytopenias or compromised stem cell activity. The possibility of depleting normal hematopoietic stem and progenitor cells with extended treatment regimens is a legitimate safety concern that needs to be thoroughly assessed in long-term animal studies and eventually human trials, even though preliminary research indicates a therapeutic window exists.186,187 Wider preclinical restrictions exacerbate these particular biology and safety issues. The majority of preclinical evidence comes from animal models and cell lines, which could not accurately represent the complexity of human disease.188 The accuracy of standard mouse models is especially questionable because species-to-species variations in the bone marrow microenvironment are substantial. The need for more physiologically appropriate models is highlighted by recent research employing humanized models, which indicates that human-specific microenvironmental variables can change the reliance of malignant cells on ILK signaling.189 Beyond them, there are yet further obstacles. It is unclear how compensatory signaling pathway activation or other potential resistance mechanisms to ILK inhibitors itself work.58 Additional optimization is needed to address pharmacokinetic issues, such as minimizing off-target effects and ensuring the best possible drug delivery to the bone marrow. Achieving consistent therapeutic responses is further complicated by the variation in ILK expression among patient populations, which may call for logical combination approaches.58
Translational strategies and future directions
Biomarker development for patient selection
To maximize the clinical impact of ILK-targeted therapies, robust biomarkers for patient selection and response monitoring are essential.190,191 Several promising biomarker strategies are under development, including ILK expression levels, phospho-protein signatures, transcriptional profiling, and functional assays.22,192 Initial studies suggest that patients with ILK expression ≥ 5-fold above normal controls may derive the greatest benefit from ILK inhibition.33,193
Preliminary validation studies suggest that high ILK expression combined with elevated phospho-Akt (Ser473) levels may identify patients most likely to respond to ILK inhibition.194,195 Ongoing research aims to refine these biomarker approaches and integrate them into planned clinical trials.196,197
Novel delivery systems for ILK inhibitors
Optimizing the delivery of ILK inhibitors to malignant cells while minimizing systemic exposure represents an important approach to enhance efficacy and reduce potential toxicities.22,198 Several innovative delivery strategies are under investigation, including nanoparticle formulations, antibody-drug conjugates, and bone marrow-targeted delivery systems.199,200
Recent innovations in nanoparticle-based drug delivery systems have shown particular promise for targeting ILK inhibitors to leukemic cells within the bone marrow microenvironment.201,202 Zhao et al developed a novel bone marrow-targeted nanoparticle system that selectively delivers ILK inhibitors to malignant cells while minimizing off-target effects.201 This approach significantly enhanced the therapeutic efficacy and safety profile of ILK inhibitors in preclinical models, suggesting potential clinical applications in the near future.201,202
Immune evasion mechanisms: The emerging role of ILK in immunotherapy resistance
An emerging area of interest is ILK's role in mediating resistance to immunotherapies, particularly checkpoint inhibitors and CAR-T cell therapy, which have revolutionized treatment for certain hematologic malignancies.158,203 Recent research has revealed that activation of ILK in malignant B cells upregulates PD-L1 expression, therefore promoting immune evasion and resistance to checkpoint inhibitor treatment.158 Particularly, the study conducted by Almasabi et al indicated that ILK expression correlates with PD-L1 expression and immune cell cytotoxicity in colorectal cancer, therefore implying its function in the tumor microenvironment and immune evasion mechanisms.
Furthermore, the study emphasizes how lowered PD-L1 expression caused by ILK gene deletion in PD-L1 positive cell lines suggests ILK might be a therapeutic target to stop immune evasion.158
In the context of CAR-T cell therapy, ILK activation in leukemic cells has been shown to enhance expression of adhesion molecules and anti-apoptotic proteins, creating a formidable barrier against CAR-T cell-mediated cytotoxicity.48 Preliminary studies suggest that combining ILK inhibitors with CAR-T cell therapy may enhance CAR-T cell efficacy and durability, potentially addressing the significant challenge of relapse following initial CAR-T cell responses.164,204
Furthermore, ILK modulates the tumor immune microenvironment by influencing cytokine production and immune cell recruitment.205 High ILK expression in AML blasts correlates with increased production of immunosuppressive cytokines such as IL-10 and TGF-β, creating an environment that inhibits natural killer cell and cytotoxic T cell functions.206 Targeting ILK may therefore have the dual benefit of sensitizing malignant cells to therapy while simultaneously enhancing anti-tumor immune responses.207,208 Overall, the central role of ILK in hematologic malignancies has been summarized in Fig. 3.
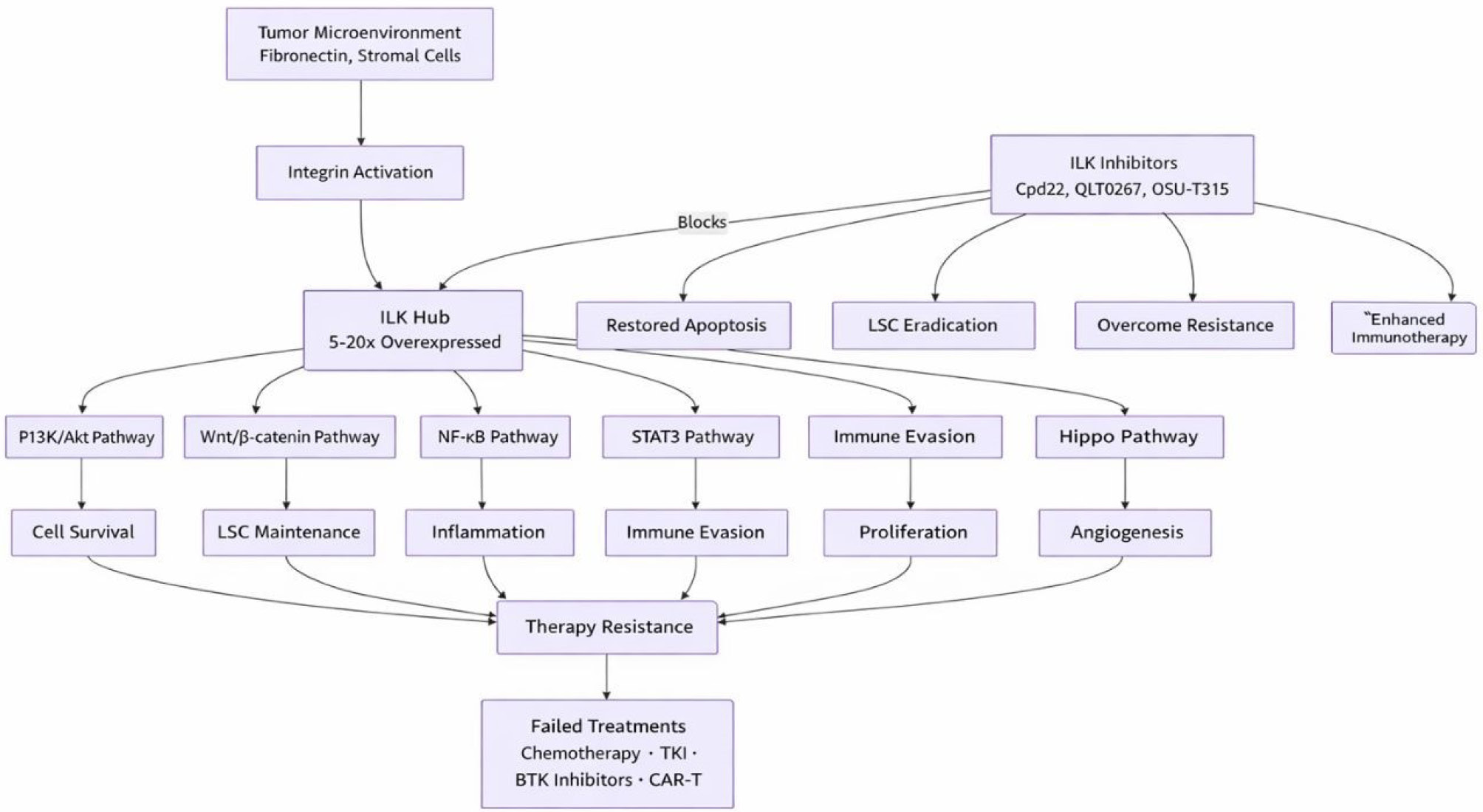
Fig. 3.
The central role of ILK in hematologic malignancies. ILK is significantly overexpressed as a result of integrin activation by the tumor microenvironment. Thereafter, ILK acts as a molecular center, coordinating several pro-survival pathways that together create a therapy-resistant state. This network is simultaneously disrupted by the deliberate suppression of ILK, which overcomes resistance and makes cancerous cells more sensitive to other therapies.
.
The central role of ILK in hematologic malignancies. ILK is significantly overexpressed as a result of integrin activation by the tumor microenvironment. Thereafter, ILK acts as a molecular center, coordinating several pro-survival pathways that together create a therapy-resistant state. This network is simultaneously disrupted by the deliberate suppression of ILK, which overcomes resistance and makes cancerous cells more sensitive to other therapies.
Conclusions
Integrin-linked kinase (ILK) has emerged as a critical mediator of oncogenic signaling and therapeutic resistance in hematologic malignancies. This comprehensive review has elucidated ILK's multifaceted roles in leukemias, lymphomas, and multiple myeloma, highlighting its potential as a therapeutic target. Several key findings and implications emerge from this analysis. ILK exhibits significantly elevated expression across diverse hematologic malignancies compared to normal counterparts, with quantitative studies demonstrating 5-20-fold increases in leukemic cells. This differential expression correlates with disease aggressiveness and treatment resistance, providing both a diagnostic marker and therapeutic opportunity. Targeting ILK exploits the higher dependency of malignant cells on this signaling hub, creating a therapeutic window that may minimize effects on normal tissues. ILK regulates multiple downstream signaling pathways essential for cancer cell survival and therapy resistance. The unique ability of ILK to integrate inputs from the tumor microenvironment and receptor signals and transduce them to multiple effector pathways makes it a strategic target with significant potential to overcome therapy resistance. Preclinical evidence suggests that targeting ILK may be particularly effective in eliminating therapy-resistant leukemic stem cells (LSCs). A major challenge in treating hematologic malignancies is the complete eradication of the LSC population, which typically resists conventional chemotherapy and targeted therapy and can drive cancer recurrence. ILK inhibitors, by interfering with the Wnt/β-catenin pathway and disrupting microenvironmental connections, selectively target LSC viability while having minimal impact on normal hematopoietic progenitors. As our understanding of ILK biology continues to evolve, and as more potent and selective inhibitors enter clinical evaluation, targeting this critical signaling hub offers an innovative approach to addressing the persistent challenge of therapy resistance in hematologic malignancies. By disrupting both intrinsic survival pathways and microenvironment-mediated protection, ILK inhibition represents a promising strategy to improve outcomes for patients with these challenging diseases.
Review Highlights
What is the current knowledge?
Existing studies suggest ILK contributes to oncogenic signaling in leukemia and lymphoma, yet mechanistic insights and therapeutic relevance are limited.
What is new here?
We evaluate emerging ILK-targeted strategies, highlighting their translational potential for precision oncology in blood cancers.
Competing Interests
There is no competing interest to be declared.
Data Availability Statement
Not applicable.
Ethical Approval
Not applicable.
References
- Wei S, Chen X, Zhang X, Chen L. Recent development of graphene based electrochemical sensor for detecting hematological malignancies-associated biomarkers: a mini-review. Front Chem 2021; 9:735668. doi: 10.3389/fchem.2021.735668 [Crossref] [ Google Scholar]
- Cui X, Zhao X, Liang Y. Sex differences in normal and malignant hematopoiesis. Blood Sci 2022; 4:185-91. doi: 10.1097/bs9.0000000000000133 [Crossref] [ Google Scholar]
- Hofmann WK, Trumpp A, Müller-Tidow C. Therapy resistance mechanisms in hematological malignancies. Int J Cancer 2023; 152:340-7. doi: 10.1002/ijc.34243 [Crossref] [ Google Scholar]
- Garcia GG, Schmidt CJ, Hajal C. The tumor microenvironment in therapy resistance. Front Lab Chip Technol 2024; 3:1420233. doi: 10.3389/frlct.2024.1420233 [Crossref] [ Google Scholar]
- Son B, Lee S, Youn H, Kim E, Kim W, Youn B. The role of tumor microenvironment in therapeutic resistance. Oncotarget 2017; 8:3933-45. doi: 10.18632/oncotarget.13907 [Crossref] [ Google Scholar]
- Park E, Chen J, Moore A, Mangolini M, Santoro A, Boyd JR. Stromal cell protein kinase C-β inhibition enhances chemosensitivity in B cell malignancies and overcomes drug resistance. Sci Transl Med 2020; 12:eaax9340. doi: 10.1126/scitranslmed.aax9340 [Crossref] [ Google Scholar]
- Miao R, Lim VY, Kothapalli N, Ma Y, Fossati J, Zehentmeier S. Hematopoietic Stem Cell Niches and Signals Controlling Immune Cell Development and Maintenance of Immunological Memory. Front Immunol 2020; 11:600127. doi: 10.3389/fimmu.2020.600127 [Crossref] [ Google Scholar]
- Khattab S, El Sorady M, El-Ghandour A, Visani G, Piccaluga PP. Hematopoietic and leukemic stem cells homeostasis: the role of bone marrow niche. Explor Target Antitumor Ther 2024; 5:1027-55. doi: 10.37349/etat.2024.00262 [Crossref] [ Google Scholar]
- Khademi R, Malekzadeh H, Bahrami S, Saki N, Khademi R, Villa-Diaz LG. Regulation and Functions of α6-Integrin (CD49f) in Cancer Biology. Cancers (Basel) 2023; 15:3466. doi: 10.3390/cancers15133466 [Crossref] [ Google Scholar]
- Ahmed AU, Almasabi S, Firestein R, Williams BRG. Integrin-linked kinase expression in myeloid cells promotes colon tumorigenesis. Front Immunol 2023; 14:1270194. doi: 10.3389/fimmu.2023.1270194 [Crossref] [ Google Scholar]
- Mehra S, Nicholls M, Taylor J. The Evolving Role of Bruton's Tyrosine Kinase Inhibitors in B Cell Lymphomas. Int J Mol Sci 2024; 25:7516. doi: 10.3390/ijms25147516 [Crossref] [ Google Scholar]
- Ondrisova L, Mraz M. Genetic and Non-Genetic Mechanisms of Resistance to BCR Signaling Inhibitors in B Cell Malignancies. Front Oncol 2020; 10:591577. doi: 10.3389/fonc.2020.591577 [Crossref] [ Google Scholar]
- Smith CIE, Burger JA. Resistance Mutations to BTK Inhibitors Originate From the NF-κB but Not From the PI3K-RAS-MAPK Arm of the B Cell Receptor Signaling Pathway. Front Immunol 2021; 12:689472. doi: 10.3389/fimmu.2021.689472 [Crossref] [ Google Scholar]
-
Pilmis B, Kherabi Y, Huriez P, Zahar JR, Mokart D. Infectious Complications of Targeted Therapies for Solid Cancers or Leukemias/Lymphomas. Cancers (Basel) 2023; 15. doi: 10.3390/cancers15071989.
- Song MK, Park BB, Uhm JE. Resistance Mechanisms to CAR T-Cell Therapy and Overcoming Strategy in B-Cell Hematologic Malignancies. Int J Mol Sci 2019; 20:5010. doi: 10.3390/ijms20205010 [Crossref] [ Google Scholar]
- Mandeville TK, Wright JP, Mavis C, Cortese M, Matthew J, Rosario S. Evasion of αCD19 CAR T-Mediated Killing By Rituximab-Chemotherapy Resistant Lymphoma Cells. Blood 2024; 144:5762. doi: 10.1182/blood-2024-212215 [Crossref] [ Google Scholar]
- Wu C, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol 2001; 155:505-10. doi: 10.1083/jcb.200108077 [Crossref] [ Google Scholar]
- Elad N, Volberg T, Patla I, Hirschfeld-Warneken V, Grashoff C, Spatz JP. The role of integrin-linked kinase in the molecular architecture of focal adhesions. J Cell Sci 2013; 126:4099-107. doi: 10.1242/jcs.120295 [Crossref] [ Google Scholar]
- Li F, Zhang Y, Wu C. Integrin-linked kinase is localized to cell-matrix focal adhesions but not cell-cell adhesion sites and the focal adhesion localization of integrin-linked kinase is regulated by the PINCH-binding ANK repeats. J Cell Sci 1999; 112:4589-99. doi: 10.1242/jcs.112.24.4589 [Crossref] [ Google Scholar]
- Tu Y, Li F, Goicoechea S, Wu C. The LIM-only protein PINCH directly interacts with integrin-linked kinase and is recruited to integrin-rich sites in spreading cells. Mol Cell Biol 1999; 19:2425-34. doi: 10.1128/mcb.19.3.2425 [Crossref] [ Google Scholar]
- Li F, Zhang Y, Wu C. Integrin-linked kinase is localized to cell-matrix focal adhesions but not cell-cell adhesion sites and the focal adhesion localization of integrin-linked kinase is regulated by the PINCH-binding ANK repeats. J Cell Sci 1999; 112(Pt 24):4589-99. doi: 10.1242/jcs.112.24.4589 [Crossref] [ Google Scholar]
- Zheng CC, Hu HF, Hong P, Zhang QH, Xu WW, He QY. Significance of integrin-linked kinase (ILK) in tumorigenesis and its potential implication as a biomarker and therapeutic target for human cancer. Am J Cancer Res 2019; 9:186-97. [ Google Scholar]
- Morgner J, Wickström SA. The weakest link: a new paradigm for stabilizing the integrin-actin connection. Cell Cycle 2013; 12:2929-30. doi: 10.4161/cc.26213 [Crossref] [ Google Scholar]
- Stanchi F, Grashoff C, Nguemeni Yonga CF, Grall D, Fässler R, Van Obberghen-Schilling E. Molecular dissection of the ILK-PINCH-parvin triad reveals a fundamental role for the ILK kinase domain in the late stages of focal-adhesion maturation. J Cell Sci 2009; 122:1800-11. doi: 10.1242/jcs.044602 [Crossref] [ Google Scholar]
- Zervas CG, Psarra E, Williams V, Solomon E, Vakaloglou KM, Brown NH. A central multifunctional role of integrin-linked kinase at muscle attachment sites. J Cell Sci 2011; 124:1316-27. doi: 10.1242/jcs.081422 [Crossref] [ Google Scholar]
- Teyssier V, Williamson CR, Shata E, Rosen SP, Jones N, Bisson N. Adapting to change: resolving the dynamic and dual roles of NCK1 and NCK2. Biochem J 2024; 481:1411-35. doi: 10.1042/bcj20230232 [Crossref] [ Google Scholar]
- Machado-Neto JA, Fenerich BA, Rodrigues Alves APN, Fernandes JC, Scopim-Ribeiro R, Coelho-Silva JL. Insulin Substrate Receptor (IRS) proteins in normal and malignant hematopoiesis. Clinics (Sao Paulo) 2018; 73:e566s. doi: 10.6061/clinics/2018/e566s [Crossref] [ Google Scholar]
- Shaw LM. Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the alpha6beta4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol Cell Biol 2001; 21:5082-93. doi: 10.1128/mcb.21.15.5082-5093.2001 [Crossref] [ Google Scholar]
- Leung-Hagesteijn C, Mahendra A, Naruszewicz I, Hannigan GE. Modulation of integrin signal transduction by ILKAP, a protein phosphatase 2C associating with the integrin-linked kinase, ILK1. Embo j 2001; 20:2160-70. doi: 10.1093/emboj/20.9.2160 [Crossref] [ Google Scholar]
- Murányi A, MacDonald JA, Deng JT, Wilson DP, Haystead TA, Walsh MP. Phosphorylation of the myosin phosphatase target subunit by integrin-linked kinase. Biochem J 2002; 366:211-6. doi: 10.1042/bj20020401 [Crossref] [ Google Scholar]
- Orlov I, Myasnikov AG, Andronov L, Natchiar SK, Khatter H, Beinsteiner B. The integrative role of cryo electron microscopy in molecular and cellular structural biology. Biol Cell 2017; 109:81-93. doi: 10.1111/boc.201600042 [Crossref] [ Google Scholar]
- Fukuda K, Bledzka K, Yang J, Perera HD, Plow EF, Qin J. Molecular basis of kindlin-2 binding to integrin-linked kinase pseudokinase for regulating cell adhesion. J Biol Chem 2014; 289:28363-75. doi: 10.1074/jbc.M114.596692 [Crossref] [ Google Scholar]
-
de la Puente P, Weisberg E, Muz B, Nonami A, Luderer M, Stone RM, et al. Identification of ILK as a novel therapeutic target for acute and chronic myeloid leukemia. Leuk Res 2015. doi: 10.1016/j.leukres.2015.09.005.
- Bilajac E, Mahmutović L, Glamočlija U, Osmanović A, Hromić-Jahjefendić A, Tambuwala MM. Curcumin Decreases Viability and Inhibits Proliferation of Imatinib-Sensitive and Imatinib-Resistant Chronic Myeloid Leukemia Cell Lines. Metabolites 2022; 13:58. doi: 10.3390/metabo13010058 [Crossref] [ Google Scholar]
- Kennedy VE, Smith CC. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front Oncol 2020; 10:612880. doi: 10.3389/fonc.2020.612880 [Crossref] [ Google Scholar]
- Lam SSY, Leung AYH. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. Int J Mol Sci 2020; 21:1537. doi: 10.3390/ijms21041537 [Crossref] [ Google Scholar]
- Short NJ, Kantarjian H, Ravandi F, Daver N. Emerging treatment paradigms with FLT3 inhibitors in acute myeloid leukemia. Ther Adv Hematol 2019; 10:2040620719827310. doi: 10.1177/2040620719827310 [Crossref] [ Google Scholar]
- Travaglini S, Gurnari C, Ottone T, Voso MT. Advances in the pathogenesis of FLT3 -mutated acute myeloid leukemia and targeted treatments. Curr Opin Oncol 2024; 36:569-76. doi: 10.1097/cco.0000000000001094 [Crossref] [ Google Scholar]
- El Fakih R, Rasheed W, Hawsawi Y, Alsermani M, Hassanein M. Targeting FLT3 Mutations in Acute Myeloid Leukemia. Cells 2018; 7:4. doi: 10.3390/cells7010004 [Crossref] [ Google Scholar]
- Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med 2014; 370:2286-94. doi: 10.1056/NEJMoa1400029 [Crossref] [ Google Scholar]
- Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med 2014; 370:2352-4. doi: 10.1056/NEJMc1402716 [Crossref] [ Google Scholar]
- Stephens DM, Byrd JC. Resistance to Bruton tyrosine kinase inhibitors: the Achilles heel of their success story in lymphoid malignancies. Blood 2021; 138:1099-109. doi: 10.1182/blood.2020006783 [Crossref] [ Google Scholar]
- Bravo-Gonzalez A, Alasfour M, Soong D, Noy J, Pongas G. Advances in Targeted Therapy: Addressing Resistance to BTK Inhibition in B-Cell Lymphoid Malignancies. Cancers (Basel) 2024; 16:3434. doi: 10.3390/cancers16203434 [Crossref] [ Google Scholar]
- Mouhssine S, Maher N, Matti BF, Alwan AF, Gaidano G. Targeting BTK in B Cell Malignancies: From Mode of Action to Resistance Mechanisms. Int J Mol Sci 2024; 25:3234. doi: 10.3390/ijms25063234 [Crossref] [ Google Scholar]
- Jain P, Wang ML. Mantle cell lymphoma in 2022-A comprehensive update on molecular pathogenesis, risk stratification, clinical approach, and current and novel treatments. Am J Hematol 2022; 97:638-56. doi: 10.1002/ajh.26523 [Crossref] [ Google Scholar]
-
Tabe Y, Jin L, Tanaka N, Andreeff M, Konopleva M. Stroma-Activated Integrin-Linked Kinase (ILK) Supports Survival of Leukemic Cells Via Stimulation of Notch-Hes Signaling: New Therapeutic Opportunities. American Society of Hematology; 2005. doi: 10.1182/blood.V106.11.2466.2466.
- De La Puente P, Weisberg E, Nonami A, Luderer MJ, Stone RM, Melo JV. Integrin-Linked Kinase a Novel Therapeutic Target for Acute and Chronic Myeloid Leukemia. Blood 2015; 126:3694. doi: 10.1182/blood.V126.23.3694.3694 [Crossref] [ Google Scholar]
- Tabe Y, Jin L, Tsutsumi-Ishii Y, Xu Y, McQueen T, Priebe W. Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow-derived stromal cells. Cancer Res 2007; 67:684-94. doi: 10.1158/0008-5472.Can-06-3166 [Crossref] [ Google Scholar]
- Chiu CF, Weng JR, Jadhav A, Wu CY, Sargeant AM, Bai LY. T315 Decreases Acute Myeloid Leukemia Cell Viability through a Combination of Apoptosis Induction and Autophagic Cell Death. Int J Mol Sci 2016; 17:1337. doi: 10.3390/ijms17081337 [Crossref] [ Google Scholar]
- Wang Y, Tang X, Zhu Y, Yang XX, Liu B. Role of interleukins in acute myeloid leukemia. Leuk Lymphoma 2023; 64:1400-13. doi: 10.1080/10428194.2023.2218508 [Crossref] [ Google Scholar]
- Hsu EC, Kulp SK, Huang HL, Tu HJ, Chao MW, Tseng YC. Integrin-linked kinase as a novel molecular switch of the IL-6-NF-κB signaling loop in breast cancer. Carcinogenesis 2016; 37:430-42. doi: 10.1093/carcin/bgw020 [Crossref] [ Google Scholar]
- Hideshima T, Anderson KC. Signaling Pathway Mediating Myeloma Cell Growth and Survival. Cancers (Basel) 2021; 13:216. doi: 10.3390/cancers13020216 [Crossref] [ Google Scholar]
- Bommert K, Bargou RC, Stühmer T. Signalling and survival pathways in multiple myeloma. Eur J Cancer 2006; 42:1574-80. doi: 10.1016/j.ejca.2005.12.026 [Crossref] [ Google Scholar]
- Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999; 93:1658-67. doi: 10.1182/blood.V93.5.1658 [Crossref] [ Google Scholar]
- Schmidmaier R, Mörsdorf K, Baumann P, Emmerich B, Meinhardt G. Evidence for cell adhesion-mediated drug resistance of multiple myeloma cells in vivo. Int J Biol Markers 2006; 21:218-22. doi: 10.1177/172460080602100404 [Crossref] [ Google Scholar]
- Zhao W, Zhang X, Zang L, Zhao P, Chen Y, Wang X. ILK promotes angiogenic activity of mesenchymal stem cells in multiple myeloma. Oncol Lett 2018; 16:1101-6. doi: 10.3892/ol.2018.8711 [Crossref] [ Google Scholar]
- Mahindra A, Cirstea D, Raje N. Novel therapeutic targets for multiple myeloma. Future Oncol 2010; 6:407-18. doi: 10.2217/fon.10.2 [Crossref] [ Google Scholar]
- Zheng C-C, Hu H-F, Hong P, Zhang Q-H, Xu WW, He Q-Y. Significance of integrin-linked kinase (ILK) in tumorigenesis and its potential implication as a biomarker and therapeutic target for human cancer. Am J Cancer Res 2019; 9:186. [ Google Scholar]
-
Evangelisti C, Chiarini F, Cappellini A, Paganelli F, Fini M, Santi S, et al. Targeting Wnt/β‐catenin and PI3K/Akt/mTOR pathways in T‐cell acute lymphoblastic leukemia. J Cell Physiol 2020. 235: 5413-28. doi: 10.1002/jcp.29429.
- Bollrath J, Greten FR. IKK/NF‐κB and STAT3 pathways: central signalling hubs in inflammation‐mediated tumour promotion and metastasis. EMBO Rep 2009; 10:1314-9. doi: 10.1038/embor.2009.243 [Crossref] [ Google Scholar]
- McDonald PC, Dedhar S. New Perspectives on the Role of Integrin-Linked Kinase (ILK) Signaling in Cancer Metastasis. Cancers (Basel) 2022; 14:3209. doi: 10.3390/cancers14133209 [Crossref] [ Google Scholar]
- Hess F, Estrugo D, Fischer A, Belka C, Cordes N. Integrin-linked kinase interacts with caspase-9 and -8 in an adhesion-dependent manner for promoting radiation-induced apoptosis in human leukemia cells. Oncogene 2007; 26:1372-84. doi: 10.1038/sj.onc.1209947 [Crossref] [ Google Scholar]
- Krenn PW, Hofbauer SW, Pucher S, Hutterer E, Hinterseer E, Denk U. ILK Induction in Lymphoid Organs by a TNFα-NF-κB-Regulated Pathway Promotes the Development of Chronic Lymphocytic Leukemia. Cancer Res 2016; 76:2186-96. doi: 10.1158/0008-5472.Can-15-3379 [Crossref] [ Google Scholar]
- Kawauchi K, Ogasawara T, Yasuyama M, Otsuka K, Yamada O. Regulation and importance of the PI3K/Akt/mTOR signaling pathway in hematologic malignancies. Anticancer Agents Med Chem 2009; 9:1024-38. doi: 10.2174/187152009789377772 [Crossref] [ Google Scholar]
- Dan HC, Antonia RJ, Baldwin AS. PI3K/Akt promotes feedforward mTORC2 activation through IKKα. Oncotarget 2016; 7:21064-75. doi: 10.18632/oncotarget.8383 [Crossref] [ Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098-101. doi: 10.1126/science.1106148 [Crossref] [ Google Scholar]
- Matheny RW, Jr Jr, Adamo ML. Current perspectives on Akt Akt-ivation and Akt-ions. Exp Biol Med (Maywood) 2009; 234:1264-70. doi: 10.3181/0904-mr-138 [Crossref] [ Google Scholar]
- Kale J, Kutuk O, Brito GC, Andrews TS, Leber B, Letai A. Phosphorylation switches Bax from promoting to inhibiting apoptosis thereby increasing drug resistance. EMBO Rep 2018; 19:e45235. doi: 10.15252/embr.201745235 [Crossref] [ Google Scholar]
- Bose P, Rahmani M, Grant S. Coordinate PI3K pathway and Bcl-2 family disruption in AML. Oncotarget 2012; 3:1499-500. doi: 10.18632/oncotarget.809 [Crossref] [ Google Scholar]
- Coloff JL, Mason EF, Altman BJ, Gerriets VA, Liu T, Nichols AN. Akt requires glucose metabolism to suppress puma expression and prevent apoptosis of leukemic T cells. J Biol Chem 2011; 286:5921-33. doi: 10.1074/jbc.M110.179101 [Crossref] [ Google Scholar]
- Linton MF, Babaev VR, Huang J, Linton EF, Tao H, Yancey PG. Macrophage Apoptosis and Efferocytosis in the Pathogenesis of Atherosclerosis. Circ J 2016; 80:2259-68. doi: 10.1253/circj.CJ-16-0924 [Crossref] [ Google Scholar]
- Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther 2005; 4:1533-40. doi: 10.1158/1535-7163.Mct-05-0068 [Crossref] [ Google Scholar]
- Cirstea D, Hideshima T, Rodig S, Santo L, Pozzi S, Vallet S. Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol Cancer Ther 2010; 9:963-75. doi: 10.1158/1535-7163.Mct-09-0763 [Crossref] [ Google Scholar]
- Ramakrishnan V, Kumar S. PI3K/AKT/mTOR pathway in multiple myeloma: from basic biology to clinical promise. Leuk Lymphoma 2018; 59:2524-34. doi: 10.1080/10428194.2017.1421760 [Crossref] [ Google Scholar]
- Demel HR, Feuerecker B, Piontek G, Seidl C, Blechert B, Pickhard A. Effects of topoisomerase inhibitors that induce DNA damage response on glucose metabolism and PI3K/Akt/mTOR signaling in multiple myeloma cells. Am J Cancer Res 2015; 5:1649-64. [ Google Scholar]
- Du J, Liu S, He J, Liu X, Qu Y, Yan W. MicroRNA-451 regulates stemness of side population cells via PI3K/Akt/mTOR signaling pathway in multiple myeloma. Oncotarget 2015; 6:14993-5007. doi: 10.18632/oncotarget.3802 [Crossref] [ Google Scholar]
- Yan H, Frost P, Shi Y, Hoang B, Sharma S, Fisher M. Mechanism by which mammalian target of rapamycin inhibitors sensitize multiple myeloma cells to dexamethasone-induced apoptosis. Cancer Res 2006; 66:2305-13. doi: 10.1158/0008-5472.Can-05-2447 [Crossref] [ Google Scholar]
- Ashihara E, Takada T, Maekawa T. Targeting the canonical Wnt/β-catenin pathway in hematological malignancies. Cancer Sci 2015; 106:665-71. doi: 10.1111/cas.12655 [Crossref] [ Google Scholar]
- Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D. β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010; 18:606-18. doi: 10.1016/j.ccr.2010.10.032 [Crossref] [ Google Scholar]
- Novak A, Dedhar S. Signaling through beta-catenin and Lef/Tcf. Cell Mol Life Sci 1999; 56:523-37. doi: 10.1007/s000180050449 [Crossref] [ Google Scholar]
- Ishii T, Furuoka H, Muroi Y, Nishimura M. Inactivation of integrin-linked kinase induces aberrant tau phosphorylation via sustained activation of glycogen synthase kinase 3beta in N1E-115 neuroblastoma cells. J Biol Chem 2003; 278:26970-5. doi: 10.1074/jbc.M304113200 [Crossref] [ Google Scholar]
- Moon RT. Wnt/β-catenin pathway. Sci STKE 2005; 2005:cm1-cm. [ Google Scholar]
- Jiang X, Mak PY, Mu H, Tao W, Mak DH, Kornblau S. Disruption of Wnt/β-Catenin Exerts Antileukemia Activity and Synergizes with FLT3 Inhibition in FLT3-Mutant Acute Myeloid Leukemia. Clin Cancer Res 2018; 24:2417-29. doi: 10.1158/1078-0432.Ccr-17-1556 [Crossref] [ Google Scholar]
- Stelmach P, Trumpp A. Leukemic stem cells and therapy resistance in acute myeloid leukemia. Haematologica 2023; 108:353-66. doi: 10.3324/haematol.2022.280800 [Crossref] [ Google Scholar]
- van Gils N, Denkers F, Smit L. Escape From Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia. Front Oncol 2021; 11:659253. doi: 10.3389/fonc.2021.659253 [Crossref] [ Google Scholar]
- Patel AB, O'Hare T, Deininger MW. Mechanisms of Resistance to ABL Kinase Inhibition in Chronic Myeloid Leukemia and the Development of Next Generation ABL Kinase Inhibitors. Hematol Oncol Clin North Am 2017; 31:589-612. doi: 10.1016/j.hoc.2017.04.007 [Crossref] [ Google Scholar]
- Chea M, Rigolot L, Canali A, Vergez F. Minimal Residual Disease in Acute Myeloid Leukemia: Old and New Concepts. Int J Mol Sci 2024; 25:2150. doi: 10.3390/ijms25042150 [Crossref] [ Google Scholar]
- Ianniciello A, Helgason GV. Targeting ULK1 in cancer stem cells: insight from chronic myeloid leukemia. Autophagy 2022; 18:1734-6. doi: 10.1080/15548627.2022.2041152 [Crossref] [ Google Scholar]
- Nishikori M. Classical and alternative NF-κB activation pathways and their roles in lymphoid malignancies. J Clin Exp Hematop 2005; 45:15-24. doi: 10.3960/jslrt.45.15 [Crossref] [ Google Scholar]
- Kennedy R, Klein U. Aberrant Activation of NF-κB Signalling in Aggressive Lymphoid Malignancies. Cells 2018; 7:189. doi: 10.3390/cells7110189 [Crossref] [ Google Scholar]
- Shaffer AL, 3rd 3rd, Phelan JD, Wang JQ, Huang D, Wright GW, Kasbekar M. Overcoming Acquired Epigenetic Resistance to BTK Inhibitors. Blood Cancer Discov 2021; 2:630-47. doi: 10.1158/2643-3230.Bcd-21-0063 [Crossref] [ Google Scholar]
- Rah B, Rather RA, Bhat GR, Baba AB, Mushtaq I, Farooq M. JAK/STAT Signaling: Molecular Targets, Therapeutic Opportunities, and Limitations of Targeted Inhibitions in Solid Malignancies. Front Pharmacol 2022; 13:821344. doi: 10.3389/fphar.2022.821344 [Crossref] [ Google Scholar]
- Ehebauer M, Hayward P, Martinez-Arias A. Notch signaling pathway. Sci STKE 2006; 2006:cm7. doi: 10.1126/stke.3642006cm7 [Crossref] [ Google Scholar]
- Joutel A, Tournier-Lasserve E. Notch signalling pathway and human diseases. Semin Cell Dev Biol 1998; 9:619-25. doi: 10.1006/scdb.1998.0261 [Crossref] [ Google Scholar]
- Venkatesh V, Nataraj R, Thangaraj GS, Karthikeyan M, Gnanasekaran A, Kaginelli SB. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig 2018; 5:5. doi: 10.21037/sci.2018.02.02 [Crossref] [ Google Scholar]
- Tabe Y, Jin L, Tsutsumi-Ishii Y, Xu Y, McQueen T, Priebe W. Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow–derived stromal cells. Cancer Res 2007; 67:684-94. doi: 10.1158/0008-5472.CAN-06-3166 [Crossref] [ Google Scholar]
- Sanchez-Martin M, Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017; 129:1124-33. doi: 10.1182/blood-2016-09-692582 [Crossref] [ Google Scholar]
- Tottone L, Zhdanovskaya N, Carmona Pestaña Á, Zampieri M, Simeoni F, Lazzari S. Histone Modifications Drive Aberrant Notch3 Expression/Activity and Growth in T-ALL. Front Oncol 2019; 9:198. doi: 10.3389/fonc.2019.00198 [Crossref] [ Google Scholar]
- Chan SM, Weng AP, Tibshirani R, Aster JC, Utz PJ. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood 2007; 110:278-86. doi: 10.1182/blood-2006-08-039883 [Crossref] [ Google Scholar]
- Evangelisti C, Chiarini F, Cappellini A, Paganelli F, Fini M, Santi S. Targeting Wnt/β-catenin and PI3K/Akt/mTOR pathways in T-cell acute lymphoblastic leukemia. J Cell Physiol 2020; 235:5413-28. doi: 10.1002/jcp.29429 [Crossref] [ Google Scholar]
- Serrano I, McDonald PC, Lock F, Muller WJ, Dedhar S. Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nat Commun 2013; 4:2976. doi: 10.1038/ncomms3976 [Crossref] [ Google Scholar]
- Park JH, Shin JE, Park HW. The Role of Hippo Pathway in Cancer Stem Cell Biology. Mol Cells 2018; 41:83-92. doi: 10.14348/molcells.2018.2242 [Crossref] [ Google Scholar]
- Mo JS, Yu FX, Gong R, Brown JH, Guan KL. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev 2012; 26:2138-43. doi: 10.1101/gad.197582.112 [Crossref] [ Google Scholar]
- Jang JW, Kim MK, Bae SC. Reciprocal regulation of YAP/TAZ by the Hippo pathway and the Small GTPase pathway. Small GTPases 2020; 11:280-8. doi: 10.1080/21541248.2018.1435986 [Crossref] [ Google Scholar]
- Zhao Y, Sheldon M, Sun Y, Ma L. New Insights into YAP/TAZ-TEAD-Mediated Gene Regulation and Biological Processes in Cancer. Cancers (Basel) 2023; 15:5497. doi: 10.3390/cancers15235497 [Crossref] [ Google Scholar]
- Mo JS, Park HW, Guan KL. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep 2014; 15:642-56. doi: 10.15252/embr.201438638 [Crossref] [ Google Scholar]
- Wei Y, Hui VLZ, Chen Y, Han R, Han X, Guo Y. YAP/TAZ: Molecular pathway and disease therapy. MedComm (2020) 2023; 4:e340. doi: 10.1002/mco2.340 [Crossref] [ Google Scholar]
- Mokhtari RB, Ashayeri N, Baghaie L, Sambi M, Satari K, Baluch N. The Hippo Pathway Effectors YAP/TAZ-TEAD Oncoproteins as Emerging Therapeutic Targets in the Tumor Microenvironment. Cancers (Basel) 2023; 15:3468. doi: 10.3390/cancers15133468 [Crossref] [ Google Scholar]
- Shi L, Wang S, Zangari M, Xu H, Cao TM, Xu C. Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget 2010; 1:22-33. doi: 10.18632/oncotarget.105 [Crossref] [ Google Scholar]
- Carvajal-Vergara X, Tabera S, Montero JC, Esparís-Ogando A, López-Pérez R, Mateo G. Multifunctional role of Erk5 in multiple myeloma. Blood 2005; 105:4492-9. doi: 10.1182/blood-2004-08-2985 [Crossref] [ Google Scholar]
- Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res 2012; 72:3228-37. doi: 10.1158/0008-5472.Can-11-3747 [Crossref] [ Google Scholar]
- Choi C, Kwon J, Lim S, Helfman DM. Integrin β1, myosin light chain kinase and myosin IIA are required for activation of PI3K-AKT signaling following MEK inhibition in metastatic triple negative breast cancer. Oncotarget 2016; 7:63466-87. doi: 10.18632/oncotarget.11525 [Crossref] [ Google Scholar]
- Ma L, Shan Y, Bai R, Xue L, Eide CA, Ou J. A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med 2014; 6:252ra121. doi: 10.1126/scitranslmed.3009073 [Crossref] [ Google Scholar]
- O'Hare T, Eide CA, Deininger MW. New Bcr-Abl inhibitors in chronic myeloid leukemia: keeping resistance in check. Expert Opin Investig Drugs 2008; 17:865-78. doi: 10.1517/13543784.17.6.865 [Crossref] [ Google Scholar]
- Chen P, Jin Q, Fu Q, You P, Jiang X, Yuan Q. Induction of Multidrug Resistance of Acute Myeloid Leukemia Cells by Cocultured Stromal Cells via Upregulation of the PI3K/Akt Signaling Pathway. Oncol Res 2016; 24:215-23. doi: 10.3727/096504016x14634208143021 [Crossref] [ Google Scholar]
- Litwińska Z, Łuczkowska K, Machaliński B. Extracellular vesicles in hematological malignancies. Leuk Lymphoma 2019; 60:29-36. doi: 10.1080/10428194.2018.1459606 [Crossref] [ Google Scholar]
- Shen Y-Q, Sun L, Wang S-M, Zheng X-Y, Xu R. Exosomal integrins in tumor progression, treatment and clinical prediction. Int J Oncol 2024; 65:1-15. doi: 10.3892/ijo.2024.5706 [Crossref] [ Google Scholar]
- Dixson AC, Dawson TR, Di Vizio D, Weaver AM. Context-specific regulation of extracellular vesicle biogenesis and cargo selection. Nature reviews Mol Cell Biol 2023; 24:454-76. doi: 10.1038/s41580-023-00576-0 [Crossref] [ Google Scholar]
- Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 2010; 10:9-22. doi: 10.1038/nrc2748 [Crossref] [ Google Scholar]
- Khalife J, Sanchez JF, Pichiorri F. Extracellular vesicles in hematological malignancies: from biomarkers to therapeutic tools. Diagnostics 2020; 10:1065. doi: 10.3390/diagnostics10121065 [Crossref] [ Google Scholar]
- Abboud ER, Coffelt SB, Figueroa YG, Zwezdaryk KJ, Nelson AB, Sullivan DE. Integrin-linked kinase: a hypoxia-induced anti-apoptotic factor exploited by cancer cells. Int J Oncol 2007; 30:113-22. doi: 10.3892/ijo.30.1.113 [Crossref] [ Google Scholar]
- Muz B, de la Puente P, Azab F, Luderer M, Azab AK. The role of hypoxia and exploitation of the hypoxic environment in hematologic malignancies. Mol Cancer Res 2014; 12:1347-54. doi: 10.1158/1541-7786.Mcr-14-0028 [Crossref] [ Google Scholar]
- Li M, Zhang D. DNA methyltransferase-1 in acute myeloid leukaemia: beyond the maintenance of DNA methylation. Ann Med 2022; 54:2011-23. doi: 10.1080/07853890.2022.2099578 [Crossref] [ Google Scholar]
- Parbin S, Kar S, Shilpi A, Sengupta D, Deb M, Rath SK. Histone deacetylases: a saga of perturbed acetylation homeostasis in cancer. J Histochem Cytochem 2014; 62:11-33. doi: 10.1369/0022155413506582 [Crossref] [ Google Scholar]
- Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem 2004; 93:57-67. doi: 10.1002/jcb.20179 [Crossref] [ Google Scholar]
- Chou CC, Chuang HC, Salunke SB, Kulp SK, Chen CS. A novel HIF-1α-integrin-linked kinase regulatory loop that facilitates hypoxia-induced HIF-1α expression and epithelial-mesenchymal transition in cancer cells. Oncotarget 2015; 6:8271-85. doi: 10.18632/oncotarget.3186 [Crossref] [ Google Scholar]
- Tseng WP, Yang SN, Lai CH, Tang CH. Hypoxia induces BMP-2 expression via ILK, Akt, mTOR, and HIF-1 pathways in osteoblasts. J Cell Physiol 2010; 223:810-8. doi: 10.1002/jcp.22104 [Crossref] [ Google Scholar]
- Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. Embo J 2000; 19:4298-309. doi: 10.1093/emboj/19.16.4298 [Crossref] [ Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE 2007; 2007:cm8. doi: 10.1126/stke.4072007cm8 [Crossref] [ Google Scholar]
- Warfel NA. Defining the mechanisms underlying cyclin dependent kinase control of HIF-1α. Oncotarget 2022; 13:454-5. doi: 10.18632/oncotarget.28208 [Crossref] [ Google Scholar]
- Zhang X, Xu Y, Liu H, Zhao P, Chen Y, Yue Z. HIF-2α-ILK Is Involved in Mesenchymal Stromal Cell Angiogenesis in Multiple Myeloma Under Hypoxic Conditions. Technol Cancer Res Treat 2018; 17:1533033818764473. doi: 10.1177/1533033818764473 [Crossref] [ Google Scholar]
- Tan C, Cruet-Hennequart S, Troussard A, Fazli L, Costello P, Sutton K. Regulation of tumor angiogenesis by integrin-linked kinase (ILK). Cancer Cell 2004; 5:79-90. doi: 10.1016/s1535-6108(03)00281-2 [Crossref] [ Google Scholar]
- Li Y, Sun R, Zou J, Ying Y, Luo Z. Dual Roles of the AMP-Activated Protein Kinase Pathway in Angiogenesis. Cells 2019; 8:752. doi: 10.3390/cells8070752 [Crossref] [ Google Scholar]
- Giallongo C, Tibullo D, Parrinello NL, La Cava P, Di Rosa M, Bramanti V. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget 2016; 7:85764-75. doi: 10.18632/oncotarget.7969 [Crossref] [ Google Scholar]
- Davis L, Recktenwald M, Hutt E, Fuller S, Briggs M, Goel A. Targeting HIF-2α in the Tumor Microenvironment: Redefining the Role of HIF-2α for Solid Cancer Therapy. Cancers (Basel) 2022; 14:1259. doi: 10.3390/cancers14051259 [Crossref] [ Google Scholar]
- Deynoux M, Sunter N, Hérault O, Mazurier F. Hypoxia and Hypoxia-Inducible Factors in Leukemias. Front Oncol 2016; 6:41. doi: 10.3389/fonc.2016.00041 [Crossref] [ Google Scholar]
- Bruno S, Mancini M, De Santis S, Monaldi C, Cavo M, Soverini S. The Role of Hypoxic Bone Marrow Microenvironment in Acute Myeloid Leukemia and Future Therapeutic Opportunities. Int J Mol Sci 2021; 22:6857. doi: 10.3390/ijms22136857 [Crossref] [ Google Scholar]
- Sakitani M. Crosstalk Between the PI3K/AKT/mTOR Signaling Pathway And The HIF-1α/CA9 Axis and its Significance in Leukemogenesis of Acute Myeloid Leukemia. International Journal of Research in Medical and Clinical Science 2024; 2:103-15. doi: 10.70829/ijrmcs.v02.i02.014 [Crossref] [ Google Scholar]
- Zhou J, Mauerer K, Farina L, Gribben JG. The role of the tumor microenvironment in hematological malignancies and implication for therapy. Front Biosci 2005; 10:1581-96. doi: 10.2741/1642 [Crossref] [ Google Scholar]
- Kwantwi LB, Rosen ST, Querfeld C. The role of signaling lymphocyte activation molecule family receptors in hematologic malignancies. Curr Opin Oncol 2024; 36:449-55. doi: 10.1097/cco.0000000000001067 [Crossref] [ Google Scholar]
- Shain KH, Landowski TH, Dalton WS. The tumor microenvironment as a determinant of cancer cell survival: a possible mechanism for de novo drug resistance. Curr Opin Oncol 2000; 12:557-63. doi: 10.1097/00001622-200011000-00008 [Crossref] [ Google Scholar]
- Popova NV, Jücker M. The Functional Role of Extracellular Matrix Proteins in Cancer. Cancers (Basel) 2022; 14:238. doi: 10.3390/cancers14010238 [Crossref] [ Google Scholar]
- Holt RU, Baykov V, Rø TB, Brabrand S, Waage A, Sundan A. Human myeloma cells adhere to fibronectin in response to hepatocyte growth factor. Haematologica 2005; 90:479-88. [ Google Scholar]
- Dolgalev I, Tikhonova AN. Connecting the Dots: Resolving the Bone Marrow Niche Heterogeneity. Front Cell Dev Biol 2021; 9:622519. doi: 10.3389/fcell.2021.622519 [Crossref] [ Google Scholar]
- Wang ZH, Ding S. [Advances in Study of Erythroblastic Island Macrophages--Review]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2025; 33:292-5. doi: 10.19746/j.cnki.issn.1009-2137.2025.01.044 [Crossref] [ Google Scholar]
- Valind A, Verhoeven BM, Enoksson J, Karlsson J, Christensson G, Mañas A. Macrophage infiltration promotes regrowth in MYCN-amplified neuroblastoma after chemotherapy. Oncoimmunology 2023; 12:2184130. doi: 10.1080/2162402x.2023.2184130 [Crossref] [ Google Scholar]
- Alchahin AM, Tsea I, Baryawno N. Recent Advances in Single-Cell RNA-Sequencing of Primary and Metastatic Clear Cell Renal Cell Carcinoma. Cancers (Basel) 2023; 15:4734. doi: 10.3390/cancers15194734 [Crossref] [ Google Scholar]
- Hirz T, Mei S, Sarkar H, Kfoury Y, Wu S, Verhoeven BM. Dissecting the immune suppressive human prostate tumor microenvironment via integrated single-cell and spatial transcriptomic analyses. Nat Commun 2023; 14:663. doi: 10.1038/s41467-023-36325-2 [Crossref] [ Google Scholar]
- Ding J, Fang Y, Zhou R, Gu Y, Du S, Lu Q. Cord-Blood Engraftment Using an Enhanced Dual-Conditioning Regimen for Malignant Hematologic Diseases. Cell Transplant 2022; 31:9636897211070238. doi: 10.1177/09636897211070238 [Crossref] [ Google Scholar]
- Kaur P, Sachs V, Christie AL, Weinstock DM, Keck JG. Characterization of new AML PDX models: Engraftment kinetics and mutational profile. Cancer Res 2018; 78:700. doi: 10.1158/1538-7445.AM2018-700 [Crossref] [ Google Scholar]
- Visconti RJ, Kolaja K, Cottrell JA. A functional three-dimensional microphysiological human model of myeloma bone disease. J Bone Miner Res 2021; 36:1914-30. doi: 10.1002/jbmr.4404 [Crossref] [ Google Scholar]
- Rodriguez C. An Overview of Organoid and 3-Dimensional Models in Multiple Myeloma. Cancer J 2021; 27:239-46. doi: 10.1097/ppo.0000000000000526 [Crossref] [ Google Scholar]
- Nefedova Y, Sullivan DM, Bolick SC, Dalton WS, Gabrilovich DI. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood 2008; 111:2220-9. doi: 10.1182/blood-2007-07-102632 [Crossref] [ Google Scholar]
- Rashad HM, Insuasti G, Almeida-Porada G, Rodriguez C. Multiple Myeloma 3D model: a platform for testing drug effects on myeloma in conjunction with the bone marrow niche. Cancer Res 2023; 83:184. doi: 10.1158/1538-7445.AM2023-184 [Crossref] [ Google Scholar]
- Quintás-Cardama A. CAR T-Cell Therapy in Large B-Cell Lymphoma. N Engl J Med 2018; 378:1065. doi: 10.1056/NEJMc1800913 [Crossref] [ Google Scholar]
- Canel M, Serrels A, Frame MC, Brunton VG. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J Cell Sci 2013; 126:393-401. doi: 10.1242/jcs.100115 [Crossref] [ Google Scholar]
- Zhang C, Zhang C, Wang H. Immune-checkpoint inhibitor resistance in cancer treatment: Current progress and future directions. Cancer Lett 2023; 562:216182. doi: 10.1016/j.canlet.2023.216182 [Crossref] [ Google Scholar]
- Almasabi S, Boyd R, Ahmed AU, Williams BRG. Integrin-Linked Kinase Expression Characterizes the Immunosuppressive Tumor Microenvironment in Colorectal Cancer and Regulates PD-L1 Expression and Immune Cell Cytotoxicity. Front Oncol 2022; 12:836005. doi: 10.3389/fonc.2022.836005 [Crossref] [ Google Scholar]
- Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z. The Mechanism of Anti-PD-L1 Antibody Efficacy against PD-L1-Negative Tumors Identifies NK Cells Expressing PD-L1 as a Cytolytic Effector. Cancer Discov 2019; 9:1422-37. doi: 10.1158/2159-8290.Cd-18-1259 [Crossref] [ Google Scholar]
- Coschi CH, Juergens RA. Overcoming Resistance Mechanisms to Immune Checkpoint Inhibitors: Leveraging the Anti-Tumor Immune Response. Curr Oncol 2023; 31:1-23. doi: 10.3390/curroncol31010001 [Crossref] [ Google Scholar]
- Kawashima S, Togashi Y. Resistance to immune checkpoint inhibitors and the tumor microenvironment. Exp Dermatol 2023; 32:240-9. doi: 10.1111/exd.14716 [Crossref] [ Google Scholar]
- Koul D, Shen R, Bergh S, Lu Y, de Groot JF, Liu TJ. Targeting integrin-linked kinase inhibits Akt signaling pathways and decreases tumor progression of human glioblastoma. Mol Cancer Ther 2005; 4:1681-8. doi: 10.1158/1535-7163.Mct-05-0258 [Crossref] [ Google Scholar]
- Mercado-Pimentel ME, Igarashi S, Dunn AM, Behbahani M, Miller C, Read CM. The Novel Small Molecule Inhibitor, OSU-T315, Suppresses Vestibular Schwannoma and Meningioma Growth by Inhibiting PDK2 Function in the AKT Pathway Activation. Austin J Med Oncol 2016; 3:1025. [ Google Scholar]
- Liu T-M, Ling Y, Phelps MA, Woyach JA, Hertlein EK, Towns WH. OSU-T315, An Integrin-Linked Kinase (ILK) Inhibitor, Induces Apoptosis By Targeting B Cell Receptor and CD49d Mediated AKT/ERK Activation In Chronic Lymphocytic Leukemia Cells. Blood 2013; 122:2523. doi: 10.1182/blood.V122.21.2523.2523 [Crossref] [ Google Scholar]
- Feriotto G, Marchetti P, Rondanin R, Tagliati F, Aguzzi S, Beninati S. Cytotoxicity of Isoxazole Curcumin Analogs on Chronic Myeloid Leukemia-Derived K562 Cell Lines Sensitive and Resistant to Imatinib. Int J Mol Sci 2023; 24:2356. doi: 10.3390/ijms24032356 [Crossref] [ Google Scholar]
- Muranyi AL, Dedhar S, Hogge DE. Targeting integrin linked kinase and FMS-like tyrosine kinase-3 is cytotoxic to acute myeloid leukemia stem cells but spares normal progenitors. Leuk Res 2010; 34:1358-65. doi: 10.1016/j.leukres.2010.01.006 [Crossref] [ Google Scholar]
- Eke I, Leonhardt F, Storch K, Hehlgans S, Cordes N. The small molecule inhibitor QLT0267 Radiosensitizes squamous cell carcinoma cells of the head and neck. PLoS One 2009; 4:e6434. doi: 10.1371/journal.pone.0006434 [Crossref] [ Google Scholar]
- Liu TM, Ling Y, Woyach JA, Beckwith K, Yeh YY, Hertlein E. OSU-T315: a novel targeted therapeutic that antagonizes AKT membrane localization and activation of chronic lymphocytic leukemia cells. Blood 2015; 125:284-95. doi: 10.1182/blood-2014-06-583518 [Crossref] [ Google Scholar]
- Yau CY, Wheeler JJ, Sutton KL, Hedley DW. Inhibition of integrin-linked kinase by a selective small molecule inhibitor, QLT0254, inhibits the PI3K/PKB/mTOR, Stat3, and FKHR pathways and tumor growth, and enhances gemcitabine-induced apoptosis in human orthotopic primary pancreatic cancer xenografts. Cancer Res 2005; 65:1497-504. doi: 10.1158/0008-5472.Can-04-2940 [Crossref] [ Google Scholar]
- Ahmed AU, Sarvestani ST, Gantier MP, Williams BR, Hannigan GE. Integrin-linked kinase modulates lipopolysaccharide- and Helicobacter pylori-induced nuclear factor κB-activated tumor necrosis factor-α production via regulation of p65 serine 536 phosphorylation. J Biol Chem 2014; 289:27776-93. doi: 10.1074/jbc.M114.574541 [Crossref] [ Google Scholar]
- Willig JB, de Couto NMG, Vianna DRB, Mariot CDS, Gnoatto SCB, Buffon A. Betulinic Acid-Brosimine B Hybrid Compound Has a Synergistic Effect with Imatinib in Chronic Myeloid Leukemia Cell Line, Modulating Apoptosis and Autophagy. Pharmaceuticals (Basel) 2023; 16:586. doi: 10.3390/ph16040586 [Crossref] [ Google Scholar]
- Radujkovic A, Topaly J, Fruehauf S, Zeller WJ. Combination treatment of imatinib-sensitive and -resistant BCR-ABL-positive CML cells with imatinib and farnesyltransferase inhibitors. Anticancer Res 2006; 26:2169-77. [ Google Scholar]
- Liu J, Zhang Y, Liu A, Wang J, Li L, Chen X. Distinct Dasatinib-Induced Mechanisms of Apoptotic Response and Exosome Release in Imatinib-Resistant Human Chronic Myeloid Leukemia Cells. Int J Mol Sci 2016; 17:531. doi: 10.3390/ijms17040531 [Crossref] [ Google Scholar]
- Gleixner KV, Herrmann H, Sadovnik I, Schuch K, Pickl WF, Sillaber C. Nilotinib and Dasatinib Produce Synergistic Growth-Inhibitory Effects In Imatinib-Resistant CML Cells, Including Subclones Bearing the Multi-Resistant BCR/ABL Mutant T315I. Blood 2010; 116:2280. doi: 10.1182/blood.V116.21.2280.2280 [Crossref] [ Google Scholar]
- Mi Z, Guo H, Wai PY, Gao C, Kuo PC. Integrin-linked kinase regulates osteopontin-dependent MMP-2 and uPA expression to convey metastatic function in murine mammary epithelial cancer cells. Carcinogenesis 2006; 27:1134-45. doi: 10.1093/carcin/bgi352 [Crossref] [ Google Scholar]
- Rothe K, Babaian A, Nakamichi N, Chen M, Watanabe A, Mager D. The focal adhesion complex Ilk-pinch-parvin is key to niche interactions and survival of quiescent stem cells in chronic myeloid leukemia in vitro and in vivo. Blood 2016; 128:936. doi: 10.1182/blood.V128.22.936.936 [Crossref] [ Google Scholar]
- Younes MN, Kim S, Yigitbasi OG, Mandal M, Jasser SA, Dakak Yazici Y. Integrin-linked kinase is a potential therapeutic target for anaplastic thyroid cancer. Mol Cancer Ther 2005; 4:1146-56. doi: 10.1158/1535-7163.Mct-05-0078 [Crossref] [ Google Scholar]
-
Dudka W, Hoser G, Mondal SS, Turos-Korgul L, Swatler J, Kusio-Kobiałka M, et al. Targeting Integrated Stress Response by ISRIB combined with imatinib attenuates STAT5 signaling and eradicates therapy-resistant Chronic Myeloid Leukemia cells. bioRxiv 2021; 2021.05. 05.442756. doi: 10.1101/2021.05.05.442756.
- Stan MR. Editorial: Renewed Excellence for 2025–2026. IEEE Transactions on Very Large Scale Integration (VLSI) Systems 2025; 33:603-26. doi: 10.1109/TVLSI.2024.3520396 [Crossref] [ Google Scholar]
- Nagano N, Hirano Y, Kimura M, Morita H, Yasukawa T. Preclinical study of novel human allogeneic adipose tissue-derived mesenchymal stem cell sheets toward a first-in-human clinical trial for myopic chorioretinal atrophy. Stem Cell Res Ther 2024; 15:498. doi: 10.1186/s13287-024-04118-z [Crossref] [ Google Scholar]
- Alarcón-Payer C, Sánchez Suárez MDM, Martín Roldán A, Puerta Puerta JM, Jiménez Morales A. Impact of Genetic Polymorphisms and Biomarkers on the Effectiveness and Toxicity of Treatment of Chronic Myeloid Leukemia and Acute Myeloid Leukemia. J Pers Med 2022; 12:1607. doi: 10.3390/jpm12101607 [Crossref] [ Google Scholar]
- Liu J, Costello PC, Pham NA, Pintillie M, Jabali M, Sanghera J. Integrin-linked kinase inhibitor KP-392 demonstrates clinical benefits in an orthotopic human non-small cell lung cancer model. J Thorac Oncol 2006; 1:771-9. doi: 10.1016/S1556-0864(15)30405-6 [Crossref] [ Google Scholar]
- Qiu S, Sheth V, Yan C, Liu J, Chacko BK, Li H. Metabolic adaptation to tyrosine kinase inhibition in leukemia stem cells. Blood 2023; 142:574-88. doi: 10.1182/blood.2022018196 [Crossref] [ Google Scholar]
- Min HY, Lee HY. Cellular Dormancy in Cancer: Mechanisms and Potential Targeting Strategies. Cancer Res Treat 2023; 55:720-36. doi: 10.4143/crt.2023.468 [Crossref] [ Google Scholar]
- Zhou H, Tan L, Liu B, Guan XY. Cancer stem cells: Recent insights and therapies. Biochem Pharmacol 2023; 209:115441. doi: 10.1016/j.bcp.2023.115441 [Crossref] [ Google Scholar]
- de la Puente P, Weisberg E, Muz B, Nonami A, Luderer M, Stone RM. Identification of ILK as a novel therapeutic target for acute and chronic myeloid leukemia. Leuk Res 2015; 39:1299-308. doi: 10.1016/j.leukres.2015.09.005 [Crossref] [ Google Scholar]
- Gullaksen S-E, Omsland M, Brevik M, Letzner J, Haugstvedt S, Gjertsen BT. CML stem cells and their interactions and adaptations to tyrosine kinase inhibitors. Leuk Lymphoma 2025; 66:1211-20. doi: 10.1080/10428194.2025.2466817 [Crossref] [ Google Scholar]
- Kalra J, Sutherland B, Stratford A, Dragowska W, Gelmon K, Dedhar S. Suppression of Her2/neu expression through ILK inhibition is regulated by a pathway involving TWIST and YB-1. Oncogene 2010; 29:6343-56. doi: 10.1038/onc.2010.366 [Crossref] [ Google Scholar]
- Lang Y, Lyu Y, Tan Y, Hu Z. Progress in construction of mouse models to investigate the pathogenesis and immune therapy of human hematological malignancy. Front Immunol 2023; 14:1195194. doi: 10.3389/fimmu.2023.1195194 [Crossref] [ Google Scholar]
- Cottrell TR, Lotze MT, Ali A, Bifulco CB, Capitini CM, Chow LQM. Society for Immunotherapy of Cancer (SITC) consensus statement on essential biomarkers for immunotherapy clinical protocols. J Immunother Cancer 2025; 13:e010928. doi: 10.1136/jitc-2024-010928 [Crossref] [ Google Scholar]
- Pavord ID, Afzalnia S, Menzies-Gow A, Heaney LG. The current and future role of biomarkers in type 2 cytokine-mediated asthma management. Clin Exp Allergy 2017; 47:148-60. doi: 10.1111/cea.12881 [Crossref] [ Google Scholar]
- Tan AC, Chan DL, Faisal W, Pavlakis N. New drug developments in metastatic gastric cancer. Therap Adv Gastroenterol 2018; 11:1756284818808072. doi: 10.1177/1756284818808072 [Crossref] [ Google Scholar]
- Tsoumas D, Nikou S, Giannopoulou E, Champeris Tsaniras S, Sirinian C, Maroulis I. ILK Expression in Colorectal Cancer Is Associated with EMT, Cancer Stem Cell Markers and Chemoresistance. Cancer Genomics Proteomics 2018; 15:127-41. doi: 10.21873/cgp.20071 [Crossref] [ Google Scholar]
- Ning Z, Zhu X, Jiang Y, Gao A, Zou S, Gu C. Integrin-Linked Kinase Is Involved In the Proliferation and Invasion of Esophageal Squamous Cell Carcinoma. J Cancer 2020; 11:324-33. doi: 10.7150/jca.33737 [Crossref] [ Google Scholar]
- Chan J, Ko FC, Yeung YS, Ng IO, Yam JW. Integrin-linked kinase overexpression and its oncogenic role in promoting tumorigenicity of hepatocellular carcinoma. PLoS One 2011; 6:e16984. doi: 10.1371/journal.pone.0016984 [Crossref] [ Google Scholar]
- Atwani R, Lazar V, Sandusky GE, Condello S. Integrin-linked kinase regulates Wnt transcriptional activity in platinum resistant ovarian cancer stem cells. Cancer Res 2023; 83:5808. doi: 10.1158/1538-7445.AM2023-5808 [Crossref] [ Google Scholar]
- Schmidmaier R, Baumann P. ANTI-ADHESION evolves to a promising therapeutic concept in oncology. Current Medicinal Chemistry 2008; 15:978-90. doi: 10.2174/092986708784049667 [Crossref] [ Google Scholar]
- Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther 2014; 14:1145-59. doi: 10.1517/14712598.2014.912270 [Crossref] [ Google Scholar]
-
Quiñones‐Díaz BI, Grafals‐Ruiz N, Barletta‐Bonanno GL, Vivas‐Mejía PE. Delivery of Interference RNA Molecules to Ovarian Cancer Cells Using Gold‐Liposome Nanoparticle Conjugates. FASEB J 2018; 32: 801.8-801.8. doi: 10.1096/fasebj.2018.32.1_supplement.801.8.
- Katsumi H, Yamashita S, Morishita M, Yamamoto A. Bone-Targeted Drug Delivery Systems and Strategies for Treatment of Bone Metastasis. Chem Pharm Bull (Tokyo) 2020; 68:560-6. doi: 10.1248/cpb.c20-00017 [Crossref] [ Google Scholar]
- Zhao X, Qi X, Liu D, Che X, Wu G. A Novel Approach for Bladder Cancer Treatment: Nanoparticles as a Drug Delivery System. Int J Nanomedicine 2024; 19:13461-83. doi: 10.2147/ijn.S498729 [Crossref] [ Google Scholar]
- Tang Q, Zhao G, Fang H, Jiang Y, Ma P, Zhou J. Nanoparticle drug delivery system for the treatment of brain tumors: Breaching the blood-brain barrier. Acta Pharm Sin B 2024; 14:2786-9. doi: 10.1016/j.apsb.2024.03.023 [Crossref] [ Google Scholar]
- Taefehshokr S, Parhizkar A, Hayati S, Mousapour M, Mahmoudpour A, Eleid L. Cancer immunotherapy: Challenges and limitations. Pathol Res Pract 2022; 229:153723. doi: 10.1016/j.prp.2021.153723 [Crossref] [ Google Scholar]
- Pérez-Moreno MA, Ciudad-Gutiérrez P, Jaramillo-Ruiz D, Reguera-Ortega JL, Abdel-Kader Martín L, Flores-Moreno S. Combined or Sequential Treatment with Immune Checkpoint Inhibitors and Car-T Cell Therapies for the Management of Haematological Malignancies: A Systematic Review. Int J Mol Sci 2023; 24:14780. doi: 10.3390/ijms241914780 [Crossref] [ Google Scholar]
- Rich AM, Hussaini HM, Parachuru VP, Seymour GJ. Toll-like receptors and cancer, particularly oral squamous cell carcinoma. Front Immunol 2014; 5:464. doi: 10.3389/fimmu.2014.00464 [Crossref] [ Google Scholar]
- Sica A, Saccani A, Bottazzi B, Polentarutti N, Vecchi A, van Damme J. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J Immunol 2000; 164:762-7. doi: 10.4049/jimmunol.164.2.762 [Crossref] [ Google Scholar]
- Thepmalee C, Panya A, Junking M, Chieochansin T, Yenchitsomanus PT. Inhibition of IL-10 and TGF-β receptors on dendritic cells enhances activation of effector T-cells to kill cholangiocarcinoma cells. Hum Vaccin Immunother 2018; 14:1423-31. doi: 10.1080/21645515.2018.1431598 [Crossref] [ Google Scholar]
- Rivas JR, Alhakeem SS, Eckenrode JM, Zhang Y, Collard JP, Hildebrandt GC. Enhancing anti-tumor immunity and responses to immune checkpoint blockade by suppressing interleukin-10 in chronic lymphocytic leukemia. Blood 2019; 134:5486. doi: 10.1182/blood-2019-127178 [Crossref] [ Google Scholar]