Bioimpacts. 16:32819.
doi: 10.34172/bi.32819
Original Article
Celastrol overcomes 5-fluorouracil resistance in osteosarcoma cells through p53-mediated apoptotic pathway modulation and P-glycoprotein inhibition: A comprehensive mechanistic study
Mohammad Ebrahimnezhad Data curation, Formal analysis, Resources, Visualization, Writing – original draft, 1, 2 
Amir Valizadeh Data curation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing, 1, 2, *
Bahman Yousefi Conceptualization, Methodology, Project administration, Supervision, 2, 3, *
Dariush Shanehbandi Methodology, Project administration, Supervision, 4
Author information:
1Student Research Committee, Tabriz University of Medical Sciences, Tabriz, Iran
2Department of Clinical Biochemistry and Laboratory Medicine, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran
3Molecular Medicine Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
4Department of Immunology, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran
Abstract
Introduction:
Osteosarcoma, a prevalent malignant bone tumor in children and adolescents, is hindered by chemoresistance, particularly to 5-fluorouracil (5-FU), driven by mechanisms such as P-glycoprotein (P-gp) overexpression and altered p53 signaling. Celastrol, a triterpenoid, exhibits anti-tumor properties, but its potential to overcome 5-FU resistance in osteosarcoma remains underexplored.
Methods:
We investigated the synergistic effects of celastrol and 5-FU in human osteosarcoma cell lines with varying p53 statuses (U-2OS, wild-type; SaOS-2, p53-null; HOS, mutant p53) using MTT assays for cytotoxicity, Chou-Talalay method for synergy, and Cell Death Detection ELISA for apoptosis. Gene expression (p53, Bax, Bcl-2, caspase-9) was quantified via qRT-PCR, P-gp levels by Western blot, and P-gp efflux activity by Rhodamine 123 assays. Non-malignant hFOB 1.19 cells served as controls.
Results:
Celastrol and 5-FU exhibited potent cytotoxicity, with combination therapy reducing IC₅₀ values 3.7- to 11.9-fold across cell lines, showing strong synergy. The combination significantly enhanced apoptosis and modulated p53-dependent (U-2OS) and -independent (SaOS-2, HOS) pathways, upregulating Bax and caspase-9 while downregulating Bcl-2. Celastrol reduced P-gp expression and increased intracellular drug accumulation, comparable to verapamil.
Conclusion:
Celastrol synergizes with 5-FU to overcome chemoresistance in osteosarcoma by enhancing p53-mediated and -independent apoptosis and inhibiting P-gp-mediated drug efflux. These findings suggest a promising low-toxicity therapeutic strategy, warranting further in vivo and clinical investigations.
Graphical Abstract
Keywords: Celastrol, 5-Fluorouracil, Osteosarcoma, Chemoresistance, Apoptosis, P-gp
Copyright and License Information
© 2026 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
The research protocol was approved and supported by the Student Research Committee, Tabriz University of Medical Sciences, Tabriz, Iran (Grant number: 74067).
Introduction
Osteosarcoma, the most prevalent primary malignant bone tumor, predominantly affects children and adolescents, characterized by aggressive local invasion and a propensity for early systemic metastases.1 Despite advancements in multi-agent, dose-intensive chemotherapy combined with refined surgical techniques, the 5-year survival rate for patients with localized osteosarcoma has plateaued at approximately 60% over the past three decades.2 This stagnation underscores the urgent need for novel therapeutic strategies to improve outcomes, particularly given the limitations of current chemotherapeutic regimens. High-dose chemotherapy, while effective, is often constrained by significant systemic toxicity, leading to severe side effects that compromise patient tolerability and quality of life.3 Standard treatments for osteosarcoma typically integrate chemotherapy, radiotherapy, surgery, and immunotherapy, with chemotherapy remaining the cornerstone of management. However, the therapeutic efficacy of conventional chemotherapeutic agents is frequently suboptimal, and their use is associated with considerable adverse effects, necessitating the exploration of alternative or adjunctive therapies that enhance efficacy while minimizing toxicity.4
Among the chemotherapeutic agents, 5-FU is widely employed across various cancers due to its ability to inhibit thymidylate synthase, thereby disrupting DNA synthesis and inducing cancer cell death.5 Although 5-FU is not a frontline component of standard osteosarcoma regimens, where platinum-based agents and anthracyclines predominate, its inclusion in preclinical models and select multi-agent protocols for osteosarcoma has provided valuable insights into pyrimidine analog resistance mechanisms that mirror those observed with core therapeutics.6 These shared pathways, including P-gp-mediated efflux and altered DNA repair, position 5-FU as an ideal probe for studying chemosensitization strategies applicable to broader osteosarcoma treatment paradigms, particularly in relapsed or resistant cases where dose intensification is limited by toxicity. Despite its efficacy, 5-FU's clinical utility in osteosarcoma is hindered by chemoresistance, often mediated by mechanisms such as P-gp) overexpression, which actively effluxes drugs from cancer cells, reducing intracellular drug accumulation.7 Additionally, the side effects of 5-FU, including gastrointestinal toxicity and myelosuppression, further limit its use in some patients. Consequently, there has been growing interest in identifying novel anticancer agents or combination therapies that can enhance the efficacy of existing drugs like 5-FU, reduce required doses, and mitigate associated toxicities.6
Celastrol, a triterpenoid derived from the root bark of Tripterygium wilfordii Hook F., commonly known as Thunder of God Vine, has emerged as a promising candidate in this context.8 This traditional Chinese medicinal compound has demonstrated potent anti-tumor effects across various cancer types by inhibiting cell proliferation, inducing apoptosis, and triggering autophagy through multiple signaling pathways, including reactive oxygen species (ROS)/c-Jun N-terminal kinase (JNK) and mitochondrial-dependent pathways.9 In osteosarcoma, celastrol has been shown to cause G2/M phase cell cycle arrest, activate caspases, caspase-3, -8, and -9, and promote both intrinsic and extrinsic apoptotic pathways, as well as autophagy, as evidenced by autophagosome formation and LC3B-II accumulation.10,11 Notably, celastrol’s ability to inhibit P-gp and other multidrug resistance–associated proteins (e.g., MRP1, BCRP) has been reported to reverse chemoresistance in various cancers, including gastric and lung cancers, enhancing the intracellular accumulation of chemotherapeutic agents.12 Furthermore, celastrol’s modulation of additional pathways, such as endoplasmic reticulum stress (ERS) and Wnt/β-catenin signaling, underscores its multifaceted anti-neoplastic potential.13
Despite these promising attributes, the specific effects of celastrol on osteosarcoma, particularly in combination with 5-FU, remain underexplored. The interplay between celastrol’s mechanisms, such as p53-mediated apoptosis and P-gp inhibition, and its potential to overcome 5-FU resistance in osteosarcoma cells with varying p53 statuses (wild-type, null, or mutant) warrants comprehensive investigation. This study aims to elucidate the synergistic effects of celastrol and 5-FU in human osteosarcoma cell lines, focusing on their modulation of p53-dependent and -independent apoptotic pathways and P-gp-mediated drug efflux. By addressing these mechanisms, we seek to provide a mechanistic foundation for the development of novel, low-toxicity combination therapies to improve outcomes for osteosarcoma patients facing the persistent challenge of chemoresistance.
Materials and Methods
Cell lines and culture conditions
Human osteosarcoma cell lines with varying p53 statuses were employed: U-2OS (wild-type p53), SaOS-2 (p53-null), and HOS (mutant p53). These lines were sourced from the American Type Culture Collection (ATCC, Manassas, VA, USA). As a non-malignant (normal) control comparator for the osteosarcoma lines, the human fetal osteoblast cell line hFOB 1.19 (ATCC) was included. These specific cell lines were selected to represent the spectrum of p53 alterations commonly observed in osteosarcoma (wild-type, null, and mutant), allowing us to dissect whether the synergistic effects of celastrol and 5-FU on apoptosis and chemoresistance reversal are p53-dependent or occur through independent pathways, thereby enhancing the translational relevance to heterogeneous patient tumors. The osteosarcoma cells were cultured in Dulbecco's Modified Eagle Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Thermo Fisher Scientific), 100 U/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific). Cultures were maintained at 37°C in a humidified incubator with 5% CO₂. The hFOB 1.19 cells were grown in a 1:1 mixture of DMEM and Ham's F-12 medium (Thermo Fisher Scientific) supplemented with 10% FBS and the same antibiotics, at 34 °C without CO₂ supplementation. All cell lines were routinely tested for mycoplasma contamination and authenticated via short tandem repeat profiling.
Reagents and drug preparation
Celastrol ( ≥ 98% purity) and 5-FU were acquired from Sigma-Aldrich (St. Louis, MO, USA). Celastrol was solubilized in dimethyl sulfoxide (DMSO; Sigma-Aldrich) to form a 50 mM stock, while 5-FU was prepared as a 100 mM stock in sterile phosphate-buffered saline (PBS; Thermo Fisher Scientific). Stocks were aliquoted and frozen at -20 °C. For experiments, drugs were freshly diluted in complete growth medium, ensuring the final DMSO concentration remained below 0.1% (v/v) to prevent solvent-induced toxicity. Verapamil and Rhodamine 123, used in functional assays, were also from Sigma-Aldrich.
Cell viability and cytotoxicity assessment
Cytotoxic effects were quantified using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Sigma-Aldrich). Cells were plated at 5 × 10³ per well in 96-well plates and incubated overnight for attachment. Treatments involved serial dilutions of celastrol (0-100 μM), 5-FU (0-50 μM), or combinations thereof, for 48 h. Combination experiments used a constant 3:1 molar ratio (celastrol:5-FU), selected from initial dose-response optimization. Post-treatment, 20 μL of MTT (5 mg/mL in PBS) was added per well, followed by a 4-h incubation at 37 °C. Formazan crystals were dissolved in 150 μL DMSO, and optical density was read at 570 nm on a Synergy H1 microplate reader (BioTek, Winooski, VT, USA). Viability was expressed relative to vehicle-treated controls. Half-maximal inhibitory concentrations (IC₅₀) were derived via nonlinear regression in GraphPad Prism v9.5 (GraphPad Software, La Jolla, CA, USA). Experiments were conducted in triplicate wells and replicated three times.
Drug synergy analysis
Synergistic interactions were evaluated using the Chou-Talalay method. Combination index (CI) values were computed with CompuSyn software (ComboSyn Inc., Paramus, NJ, USA), where CI < 0.9 denotes synergy, CI = 0.9-1.1 additivity, and CI > 1.1 antagonism. Dose reduction indices (DRIs) were also calculated to quantify reductions in individual drug doses needed for equivalent efficacy in combination. Data from MTT assays at multiple effect levels (e.g., ED₅₀) were analyzed from at least three independent runs.
Apoptosis detection
Apoptotic DNA fragmentation was measured with the Cell Death Detection ELISA PLUS kit (Roche Diagnostics, Mannheim, Germany), following the manufacturer's guidelines. Cells were seeded at 1 × 10⁵ per well in 6-well plates, treated with celastrol (10 μM), 5-FU (2 μM), or the combination for 48 h, then lysed. Cytoplasmic histone-associated DNA fragments were quantified spectrophotometrically at 405 nm (reference: 490 nm) using the microplate reader. Results were normalized to untreated controls and reported as fold changes. Triplicate measurements were performed across three biological replicates.
RNA extraction and quantitative real-time PCR (qRT -PCR)
Total RNA was isolated from treated cells (48 h combination exposure) using the TRIzol reagent (Invitrogen, Thermo Fisher Scientific) per protocol. RNA integrity was verified on a NanoDrop One spectrophotometer (Thermo Fisher Scientific), and 1 μg was reverse-transcribed into cDNA with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Quantitative real-time PCR (qRT-PCR) was carried out on a QuantStudio 7 Flex system (Applied Biosystems) using PowerUp SYBR Green Master Mix (Applied Biosystems). Primers were:
-
p53: Forward 5'-CCCCTCCTGGCCCCTGTCATCT TC-3', Reverse 5'-GCAGCGCCTCACAACCTCCGT CAT-3'
-
Bax: Forward 5'-TGGCAGCTGACATGTTTTCTGA C-3', Reverse 5'-TCACCCAACCACCCTGGTCTT-3'
-
Bcl-2: Forward 5'-CCTGTGGATGACTGAGTAC CTG-3', Reverse 5'-GAGACAGCCAGGAGAAATC AA-3'
-
Caspase-9: Forward 5'-TGTCGCGGACTCAACTC CA-3', Reverse 5'-GCTCCTTTCACCGAAACAGC-3'
-
GAPDH: Forward 5'-ACAACTTTGGTATCGTGGA AGG-3', Reverse 5'-GCCATCACGCCACAGTTTC-3' Relative expression was determined by the 2-ΔΔCt method, normalized to GAPDH. Each sample was run in triplicate, with experiments repeated thrice.
Western blot analysis
For Western blotting, cells treated for 24 h were harvested in RIPA lysis buffer (Thermo Fisher Scientific) supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Protein was quantified via BCA assay (Pierce, Thermo Fisher Scientific). Lysates (40 μg) were resolved on 10% SDS-PAGE gels and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). Membranes were blocked in 5% bovine serum albumin in TBST (Tris-buffered saline with 0.1% Tween-20) for 1 h, then probed overnight at 4°C with primary antibodies: anti-P-glycoprotein (1:1000; ab170904, Abcam, Cambridge, UK) and anti-β-actin (1:5000; A5441, Sigma-Aldrich). Secondary HRP-linked antibodies (1:3000; Cell Signaling Technology, Danvers, MA, USA) were applied for 1 h. Bands were detected with SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific) on a ChemiDoc imaging system (Bio-Rad). Densitometry used Image Lab software (Bio-Rad), with β-actin for normalization. Assays were replicated three times.
P-glycoprotein (P- gp) efflux activity assay
P-gp function was assessed via Rhodamine 123 accumulation. After 24 h pretreatment with celastrol (10 μM), 5-FU (2 μM), or combination, cells were loaded with 5 μM Rhodamine 123 for 1 h at 37 °C, with or without 50 μM verapamil as positive control. Following washes in cold PBS, intracellular fluorescence was quantified by flow cytometry on a BD FACSCanto II (BD Biosciences, San Jose, CA, USA), analyzing 10,000 events per sample in the FITC channel. Data were processed with FlowJo v10 (BD Biosciences) and expressed as fold increase in mean fluorescence intensity over controls. Three independent experiments were performed.
Statistical analysis
Data are presented as mean ± standard deviation (SD) from a minimum of three independent experiments. Group differences were analyzed by one-way ANOVA with Tukey's post-hoc test for multiple comparisons, or two-tailed Student's t-test for pairwise analyses, using GraphPad Prism v9.5. Significance was defined as P < 0.05.
Results
Celastrol displays strong cytotoxic effects on human osteosarcoma cells
To determine the fundamental cytotoxic characteristics, we assessed the standalone impacts of celastrol and 5-FU on three distinct human osteosarcoma cell lines with varying p53 profiles: U-2OS (wild-type p53), SaOS-2 (p53-null), and HOS (mutant p53). We also included normal human osteoblasts (hFOB) for comparison. Using MTT assays, both agents exhibited cytotoxicity that increased with concentration in all cell lines examined (Fig. 1A-C).
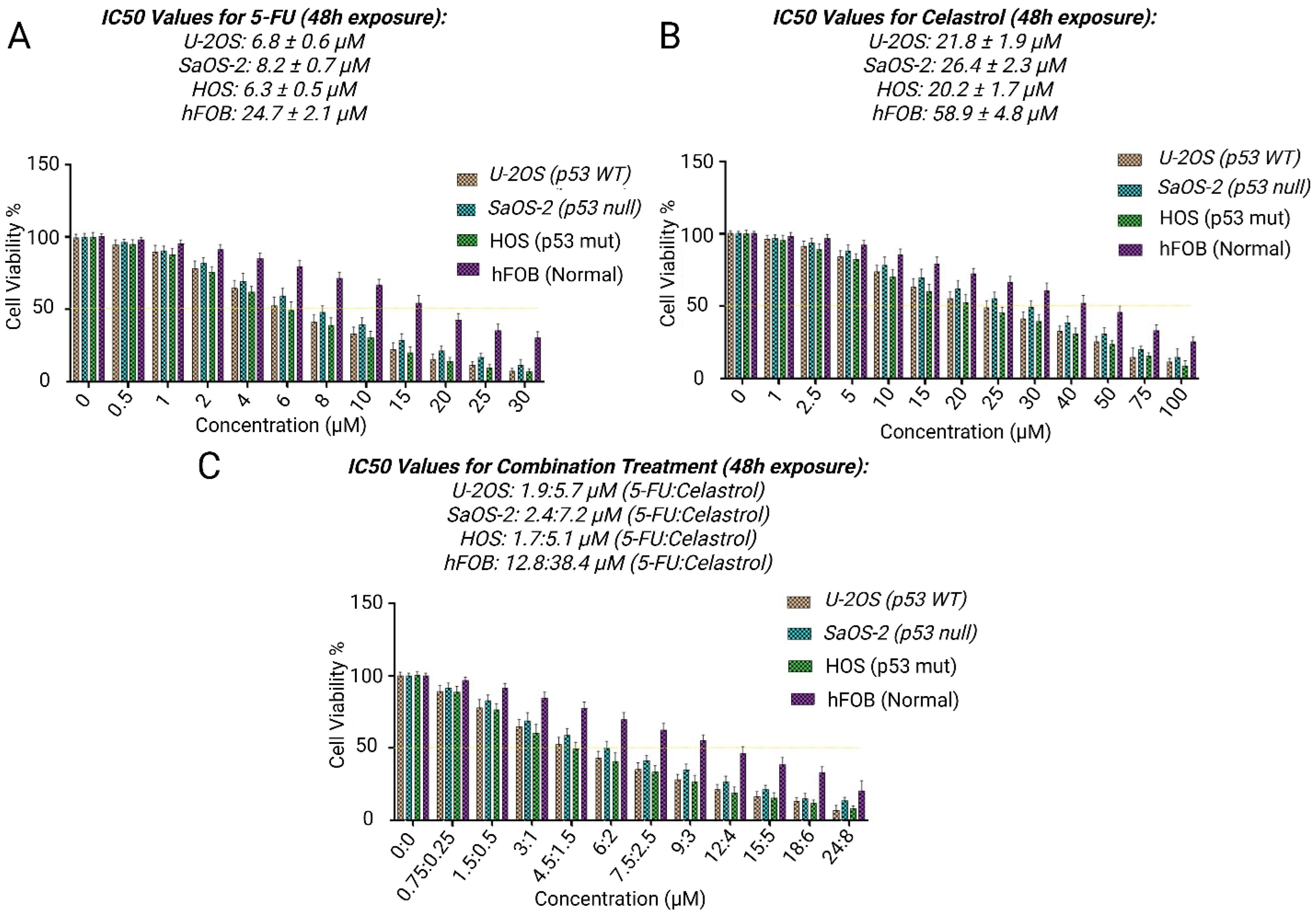
Fig. 1.
Synergistic cytotoxicity of celastrol and 5-FU alone or in combination in U-2OS, SaOS-2, HOS, and hFOB. (A) Dose-response curves showing cell viability (%) versus drug concentration (μM) for 5-FU, (B) Celastrol, and (C), combination for 48h. Data represent mean ± SD from three independent experiments performed in triplicate. ***P < 0.001 vs. individual treatments.
.
Synergistic cytotoxicity of celastrol and 5-FU alone or in combination in U-2OS, SaOS-2, HOS, and hFOB. (A) Dose-response curves showing cell viability (%) versus drug concentration (μM) for 5-FU, (B) Celastrol, and (C), combination for 48h. Data represent mean ± SD from three independent experiments performed in triplicate. ***P < 0.001 vs. individual treatments.
Celastrol exhibited robust inhibition of cell proliferation, achieving IC₅₀ values of 20.2 ± 1.7 μM for HOS cells, 24.8 ± 2.1 μM for U-2OS cells, and 26.4 ± 2.3 μM for SaOS-2 cells following 48 hours of exposure. Importantly, celastrol displayed notable preference for tumor cells, as hFOB cells were considerably more resilient (IC₅₀: 58.9 ± 4.8 μM; P < 0.001 compared to cancer lines). This translated to selectivity ratios of 2.2- to 2.9-fold, highlighting its targeted action against cancerous cells.
In parallel, 5-FU induced cytotoxicity in a dose-related manner, with IC₅₀ values of 6.3 ± 0.5 μM (HOS), 7.8 ± 0.6 μM (U-2OS), and 8.2 ± 0.7 μM (SaOS-2). The hFOB cells showed greater tolerance to 5-FU (IC₅₀: 24.7 ± 2.1 μM), resulting in selectivity ratios from 3.0- to 3.9-fold. Of note, HOS cells with mutant p53 were most responsive to each drug alone, implying that p53 alterations could affect susceptibility to these treatments.
Combined celastrol and 5-FU therapy shows robust synergism
Combination therapy at a fixed 3:1 molar ratio (celastrol:5-FU) markedly enhanced cytotoxicity across all osteosarcoma cell lines, as illustrated by the dose-response curves for 5-FU (Fig. 1A), celastrol (Fig. 1B), and the combination (Fig. 1C), with IC₅₀ values for the combination (expressed as celastrol equivalent concentration) dropping substantially compared to monotherapies (Table 1). This resulted in dose reductions of 3.7- to 11.9-fold relative to celastrol alone and 1.1- to 3.7-fold relative to 5-FU alone. Chou-Talalay analysis confirmed strong synergism (CI < 0.9 at ED₅₀ across lines), with the most pronounced effects in HOS cells (mutant p53), suggesting that p53 status may influence interaction strength.
Table 1.
Synergistic cytotoxicity and dose reduction effects of celastrol and 5-FU combination therapy in osteosarcoma cell lines with varying p53 statuses
|
Cell line
|
p53 status
|
IC₅₀ celastrol alone (μM)
|
IC₅₀ 5-FU Alone (μM)
|
IC₅₀ Combination (celastrol equivalent, μM)
|
DRI vs. celastrol alone (fold)
|
DRI vs. 5-FU alone (fold)
|
CI at ED₅₀
|
| HOS |
Mutant |
20.2 ± 1.7 |
6.3 ± 0.5 |
1.7 ± 0.2 |
11.9 |
3.7 |
0.31 ± 0.05 |
| U-2OS |
Wild-type |
24.8 ± 2.1 |
7.8 ± 0.6 |
4.9 ± 0.4 |
5.1 |
1.6 |
0.58 ± 0.08 |
| SaOS-2 |
Null |
26.4 ± 2.3 |
8.2 ± 0.7 |
7.2 ± 0.6 |
3.7 |
1.1 |
0.74 ± 0.09 |
*Notes: IC₅₀ values represent mean ± SD from three independent experiments (triplicate wells). Combination IC₅₀ is the celastrol concentration in the 3:1 (celastrol:5-FU) fixed-ratio mixture achieving 50% inhibition. DRIs were calculated using CompuSyn software as the ratio of monotherapy IC₅₀ to the effective dose in combination. CI < 0.9 indicates synergism. DRI, dose reduction index.
Dual therapy triggers extensive apoptosis
To uncover the basis for heightened toxicity, we conducted thorough apoptosis evaluations using the biochemical Cell Death Detection ELISA assay. The Cell Death Detection ELISA, measuring histone-linked DNA fragments in the cytoplasm as an apoptosis indicator, showed substantial rises in cell death after combined exposure (Fig. 2A-C).
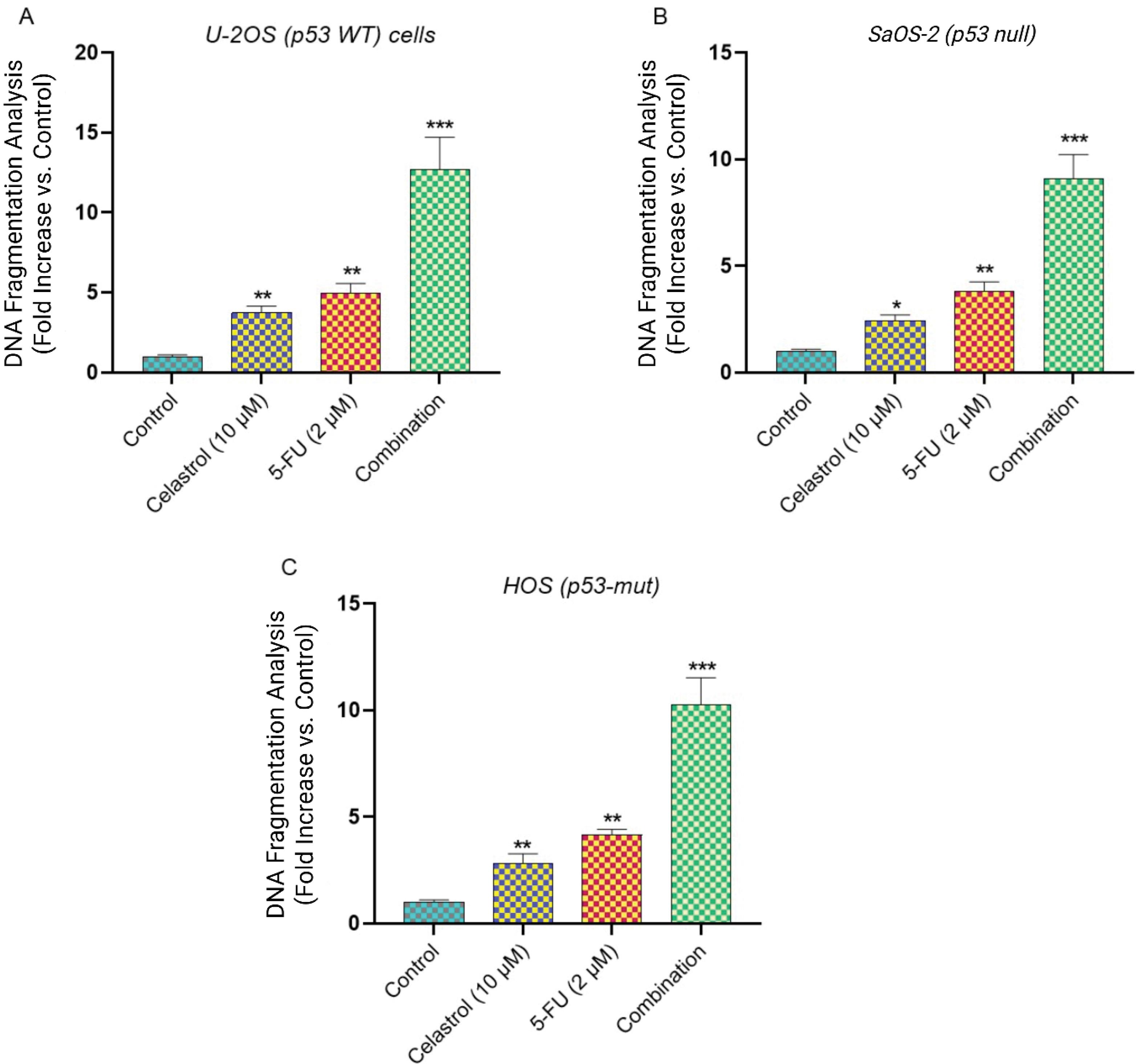
Fig. 2.
Cell death ELISA and apoptotic DNA fragmentation analysis. Quantitative analysis of DNA fragmentation using Cell Death Detection ELISA kit in (A) U-2OS, (B) SaOS-2, and (C)HOS cells treated with vehicle control, Celastrol (10 μM), 5-FU (2 μM), or combination for 48h. Data represent mean ± SD from three independent experiments. *P < 0.05 and **P < 0.01 vs. control; ***P < 0.001 vs. individual treatments.
.
Cell death ELISA and apoptotic DNA fragmentation analysis. Quantitative analysis of DNA fragmentation using Cell Death Detection ELISA kit in (A) U-2OS, (B) SaOS-2, and (C)HOS cells treated with vehicle control, Celastrol (10 μM), 5-FU (2 μM), or combination for 48h. Data represent mean ± SD from three independent experiments. *P < 0.05 and **P < 0.01 vs. control; ***P < 0.001 vs. individual treatments.
Single-agent exposures to celastrol (10 μM) or 5-FU (2 μM) over 48 hours caused moderate yet significant apoptosis over vehicle-treated groups. Celastrol alone elevated DNA fragmentation 2.8 ± 0.3-fold (U-2OS), 2.1 ± 0.2-fold (SaOS-2), and 3.4 ± 0.4-fold (HOS) versus controls (P < 0.05 to P < 0.01). 5-FU monotherapy yielded 2.2 ± 0.3-fold (U-2OS), 1.8 ± 0.2-fold (SaOS-2), and 2.9 ± 0.3-fold (HOS) increments.
In contrast, the combination elicited profound apoptotic surges, with fragmentation at 8.7 ± 0.9-fold (U-2OS), 6.2 ± 0.7-fold (SaOS-2), and 11.4 ± 1.2-fold (HOS) above baselines (P < 0.001 versus monotherapies). This underscores synergistic apoptosis promotion, particularly strong in HOS cells.
p53 condition affects apoptosis signaling
Real-time quantitative PCR uncovered unique apoptotic gene profiles linked to p53 functionality in the lines (Fig. 3A). In wild-type p53 U-2OS cells, the combination boosted p53 mRNA 4.8 ± 0.5-fold over controls (P< 0.001), alongside elevated pro-apoptotic Bax (3.2 ± 0.4-fold) and caspase-9 (2.9 ± 0.3-fold), and reduced anti-apoptotic Bcl-2 (0.3 ± 0.05-fold; P < 0.001).
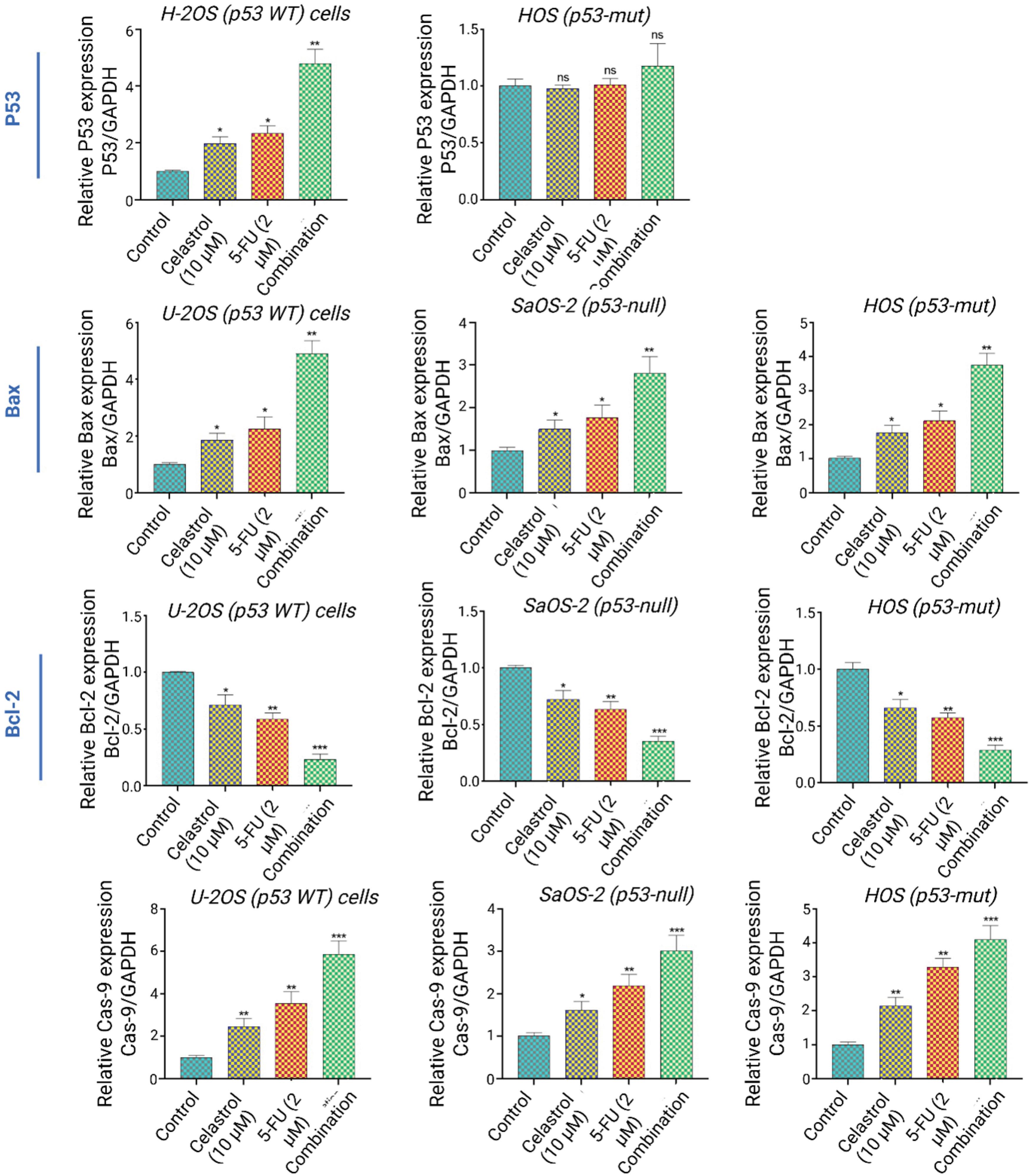
Fig. 3.
p53-Dependent and independent apoptotic pathway modulation. (A) Representative mRNA expression of p53, Bax, Bcl-2, and caspase-9 in U-2OS (p53-WT), SaOS-2 (p53-null), and HOS (p53-mut) cells following 48h combination treatment. GAPDH serves as internal control.
.
p53-Dependent and independent apoptotic pathway modulation. (A) Representative mRNA expression of p53, Bax, Bcl-2, and caspase-9 in U-2OS (p53-WT), SaOS-2 (p53-null), and HOS (p53-mut) cells following 48h combination treatment. GAPDH serves as internal control.
Conversely, p53-null SaOS-2 cells lacked p53 mRNA but activated apoptosis independently, with Bax rising 2.1 ± 0.3-fold, caspase-9 1.9 ± 0.2-fold, and Bcl-2 dropping to 0.5 ± 0.08-fold. This suggests the therapy engages both p53-reliant and autonomous routes.
Mutant p53 HOS cells presented a hybrid profile: slight p53 increase (1.8 ± 0.2-fold), strong Bax elevation (2.8 ± 0.4-fold), intermediate caspase-9 (2.3 ± 0.3-fold), and Bcl-2 decline (0.4 ± 0.06-fold). Patterns imply partial mutant p53 activity, with dominant alternative mechanisms. To more precisely evaluate the propensity for programmed cell death, we computed the Bax/Bcl-2 ratio, a reliable measure of mitochondrial susceptibility to apoptosis. Across every cell type, the combined celastrol and 5-FU regimen substantially amplified this ratio, peaking in U-2OS cells at 10.7 ± 1.3 times baseline (P < 0.001), then HOS at 7.0 ± 0.9-fold (P < 0.001), and SaOS-2 at 4.2 ± 0.6-fold (P < 0.001). Such results underscore how this synergistic approach decisively favors pro-apoptotic signaling, independent of p53 integrity.
Celastrol suppresses P- gp and boosts intracellular drug levels
Exploring synergism drivers, we analyzed celastrol's influence on P-gp levels and activity. Western blots indicated celastrol markedly lowered P-gp in all lines (Fig. 4A).
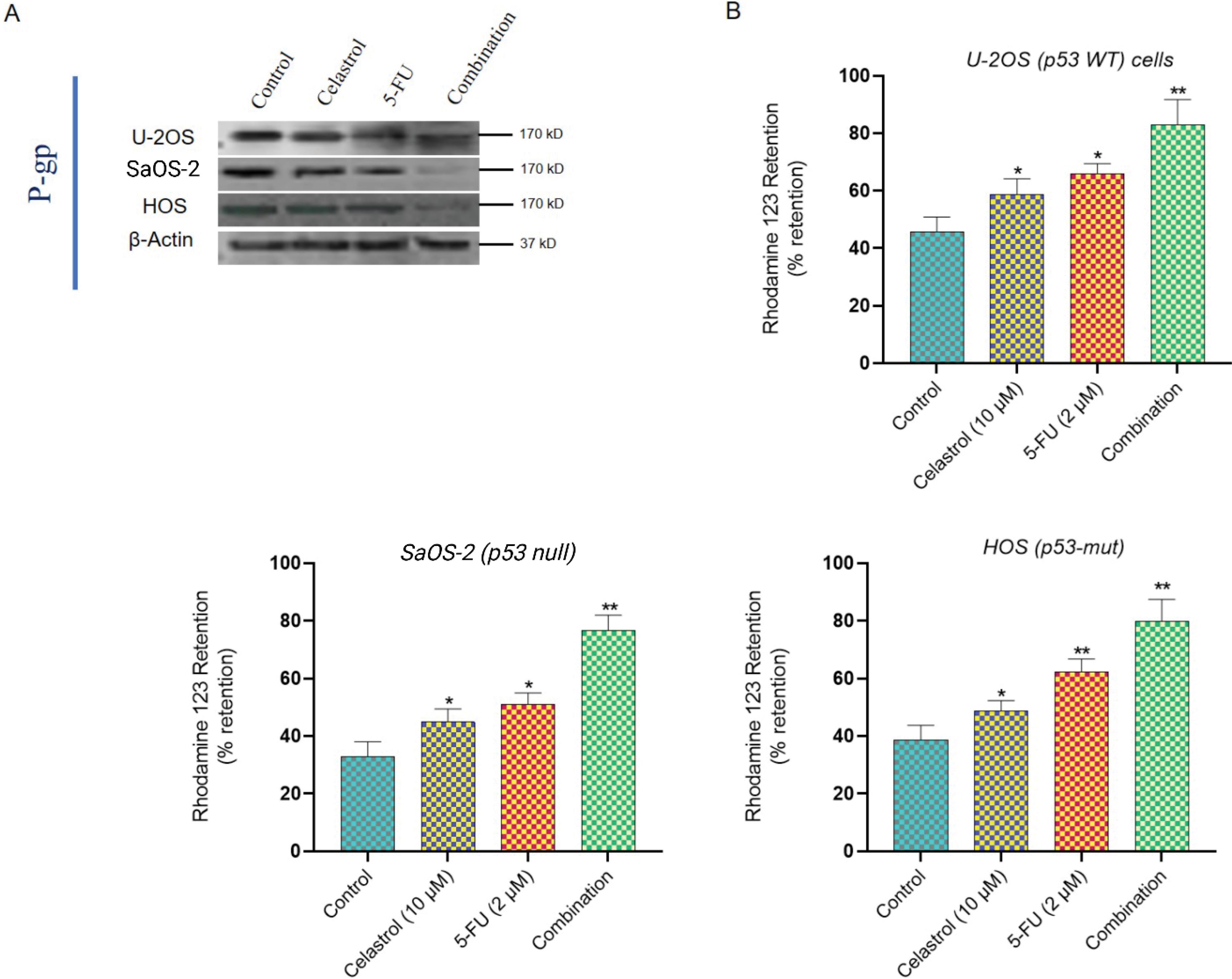
Fig. 4.
P-glycoprotein inhibition and drug accumulation enhancement. (A) Western blot analysis of P-gp protein expression in control and mono/combination-treated osteosarcoma cells. (B) Flow cytometry analysis of Rhodamine 123 retention assay demonstrating P-gp functional inhibition. Data represent mean ± SD from three independent experiments.
.
P-glycoprotein inhibition and drug accumulation enhancement. (A) Western blot analysis of P-gp protein expression in control and mono/combination-treated osteosarcoma cells. (B) Flow cytometry analysis of Rhodamine 123 retention assay demonstrating P-gp functional inhibition. Data represent mean ± SD from three independent experiments.
Initial P-gp varied, highest in SaOS-2, then HOS and U-2OS. Celastrol (10 μM, 24 hours) cut P-gp by 67 ± 8% (U-2OS), 72 ± 9% (SaOS-2), and 59 ± 7% (HOS) from controls (P < 0.001). 5-FU alone minimally affected it (15-20% drop), while combination mirrored celastrol's impact.
Rhodamine 123 assays confirmed reduced efflux (Fig. 4B). Controls had quick dye expulsion and low retention, but celastrol raised accumulation 3.8 ± 0.4-fold (U-2OS), 4.7 ± 0.5-fold (SaOS-2), and 3.2 ± 0.3-fold (HOS; P < 0.001). This matched verapamil (50 μM), a standard inhibitor, with gains of 4.1 ± 0.5-fold (U-2OS), 5.2 ± 0.6-fold (SaOS-2), and 3.5 ± 0.4-fold (HOS). Thus, celastrol hinders P-gp efflux, likely aiding 5-FU buildup and potency.
Discussion
Osteosarcoma remains a formidable challenge in pediatric oncology, with chemoresistance, particularly efflux-mediated by P-gp, contributing to stagnant survival rates despite multi-agent regimens centered on doxorubicin, cisplatin, and methotrexate. While 5-FU is not a standard component of these frontline protocols, its inclusion in this study is justified as a targeted model for investigating P-gp-driven resistance, a mechanism that impairs nucleoside analogs and broader chemotherapeutics in bone tumors. Preclinical evidence supports 5-FU's exploration in osteosarcoma for multi-drug combinations and as a probe for sensitization strategies allowing us to dissect adjunctive therapies that could enhance efficacy while minimizing toxicity across resistance phenotypes.14,15 The present study provides compelling evidence that celastrol, a triterpenoid extracted from Tripterygium wilfordii,16 significantly enhances the anti-tumor efficacy of 5-FU against human osteosarcoma cells through dual mechanisms: modulation of p53-dependent apoptotic pathways and inhibition of P-gp–mediated drug efflux. These findings address a critical clinical problem, chemoresistance, that has long hindered improvements in osteosarcoma patient survival.4 By dissecting the cooperative effects of celastrol and 5-FU in osteosarcoma cell lines with distinct p53 statuses, we highlight both p53-mediated and p53-independent apoptotic signaling as key contributors to this synergy.
Prior studies have established celastrol's anti-osteosarcoma activity primarily through monotherapy mechanisms, such as suppression of invasion and migration via downregulation of the PI3K/Akt/NF-κB pathway in U-2OS cells,17 or induction of apoptosis and autophagy via ROS/JNK signaling, with in vivo xenograft validation.18 These works underscore celastrol's broad cytotoxic potential but focus on single pathways without addressing chemoresistance reversal or combination synergies specific to osteosarcoma. Similarly, celastrol's pairing with cisplatin has been shown to amplify apoptosis via mitochondrial and endoplasmic reticulum pathways in U-2OS cells,13 yet this synergy was limited to wild-type p53 contexts and did not interrogate efflux mechanisms or p53 heterogeneity. In contrast, our study is the first to demonstrate celastrol's synergistic interaction with 5-FU, a nucleoside analog relevant to resistance modeling, in osteosarcoma across p53-wild-type (U-2OS), null (SaOS-2), and mutant (HOS) lines, revealing a distinct contribution: integrated p53-dependent/independent apoptosis modulation coupled with P-gp inhibition as dual axes for overcoming efflux-driven resistance. This extends beyond gastric cancer models, where celastrol + 5-FU synergy reduced IC50 values and induced S-phase arrest via apoptosis,19 by adapting the approach to osteosarcoma's unique genetic landscape and emphasizing P-gp downregulation (59–72% reduction) as a novel bridge to enhanced 5-FU accumulation.
Our data demonstrated that celastrol alone exerted robust cytotoxicity across all tested osteosarcoma cell lines while sparing normal osteoblasts, consistent with previous reports of its selective anti-tumor activity.20 Celastrol has been shown to trigger G2/M cell-cycle arrest, induce caspase activation, and engage both intrinsic and extrinsic apoptotic pathways in osteosarcoma and other malignancies. The present study extends these findings by showing that celastrol not only induces apoptosis directly but also sensitizes resistant cells to 5-FU. The marked reduction in combination IC₅₀ values and the low CI scores across multiple effect levels confirm a strong synergistic interaction, particularly in HOS cells harboring mutant p53. This is consistent with prior observations that celastrol can restore chemosensitivity in drug-resistant gastric and oral cancers through apoptosis induction and efflux pump inhibition.18
The apoptotic profiles we observed underscore the centrality of p53 in mediating the celastrol–5-FU interaction. In U-2OS cells with wild-type p53, combination treatment upregulated p53 mRNA nearly fivefold, accompanied by substantial increases in Bax and caspase-9 expression and a pronounced decrease in Bcl-2 levels. These molecular events align with canonical p53 signaling, which transcriptionally activates Bax and represses Bcl-2 to promote mitochondrial outer membrane permeabilization and cytochrome c release. Caspase-9 activation, observed here and in previous celastrol studies,21 further supports engagement of the intrinsic pathway. Interestingly, SaOS-2 cells, which lack functional p53, still underwent significant apoptosis under combination therapy, albeit at a reduced magnitude. Bax and caspase-9 activation in these cells indicates that celastrol and 5-FU can engage alternative pro-apoptotic mechanisms independent of p53. Such redundancy may underlie the broad effectiveness of celastrol across p53-mutant or -deficient cancers and is a particularly relevant finding given the high frequency of p53 alterations in osteosarcoma.
The intermediate response observed in HOS cells, which harbor mutant p53, suggests partial restoration or bypass of p53 signaling. Mutant p53 proteins can exert dominant-negative effects or acquire oncogenic functions that disrupt apoptosis, yet the combination therapy elicited robust apoptotic responses in this line. This suggests that celastrol may either partially reactivate mutant p53 conformations or stimulate parallel pathways, such as stress-activated kinases (e.g., JNK) and endoplasmic reticulum stress responses, both of which have been implicated in celastrol-induced apoptosis.22,23 Here too, the apoptotic marker shifts could reflect direct effects of celastrol on JNK phosphorylation, leading to secondary Bax translocation. or indirect amplification through enhanced 5-FU retention.18 Future studies employing mutant p53 rescue constructs could clarify these dynamics. The apoptotic profiles underscore p53's central role in celastrol–5-FU synergy, underexplored in prior celastrol-osteosarcoma studies lacking p53-null/mutant models. In wild-type p53 U-2OS cells, combination therapy upregulated p53 mRNA ~5-fold, boosting Bax/caspase-9 while reducing Bcl-2, consistent with canonical p53-driven mitochondrial permeabilization and intrinsic apoptosis.17 p53-null SaOS-2 cells showed reduced but significant apoptosis via Bax/caspase-9, revealing p53-independent paths that broaden celastrol's utility in p53-altered osteosarcoma. Mutant p53 HOS cells exhibited intermediate responses, implying partial p53 bypass via JNK/ER stress pathways19; shifts likely arise from JNK-mediated Bax translocation or enhanced 5-FU retention. Future mutant p53 rescue experiments could refine this, building on our p53-stratified model. A second critical mechanism contributing to the synergy is P-glycoprotein inhibition.24 P-gp overexpression is a hallmark of multidrug resistance in osteosarcoma and other tumors, actively exporting chemotherapeutics such as 5-FU out of cells and reducing intracellular drug accumulation.25,26 Our Western blot and Rhodamine 123 efflux assays revealed that celastrol substantially downregulated P-gp expression and impaired its transport activity across all osteosarcoma cell lines. Notably, the degree of P-gp inhibition by celastrol was comparable to verapamil, a standard P-gp inhibitor, and resulted in marked increases in intracellular dye retention. These findings are supported by previous studies demonstrating celastrol’s capacity to reverse multidrug resistance in gastric and lung cancer cells by suppressing P-gp and related transporters.19,27 By inhibiting P-gp, celastrol likely enhances 5-FU retention and bioavailability within osteosarcoma cells, thereby potentiating its cytotoxic effects. The pronounced synergy observed in HOS cells may be partly attributed to their relatively high baseline P-gp levels, which were significantly suppressed by celastrol. The downregulation of P-gp observed in our study could be a direct effect, as celastrol has been reported to bind the nucleotide-binding domains of P-gp, competitively inhibiting ATP hydrolysis and efflux function without altering transcription.12,27 Alternatively, it may be secondary to celastrol's broader impact on proteasomal degradation pathways or NF-κB signaling, which transcriptionally regulate MDR1 expression.28 The rapid time frame of our assays (24 h) favors a direct inhibitory mechanism over transcriptional changes, but this distinction remains tentative without additional profiling. Moreover, while our functional efflux data strongly implicate P-gp in the synergy, the lack of rescue experiments, such as stable P-gp overexpression in these cell lines to test if it attenuates celastrol's chemosensitizing effects, limits definitive causal attribution. Such experiments, alongside siRNA-mediated P-gp knockdown, would be essential to confirm P-gp as the primary mediator of enhanced 5-FU accumulation and apoptosis in future work. The reduced dose requirements for both celastrol and 5-FU under combination therapy, as indicated by favorable dose reduction indices, suggest potential clinical advantages, including lower toxicity and improved therapeutic windows. Given the dose-limiting adverse effects of both celastrol and 5-FU in vivo, this finding is particularly promising for translation into clinical strategies.
In addition to its effects on P-gp and p53 signaling, celastrol has been reported to modulate multiple cancer-related pathways, including ROS/JNK signaling, ER stress, mTOR inhibition, and Wnt/β-catenin suppression.29-31 While these pathways were not directly interrogated in the present study, their documented roles in celastrol-mediated apoptosis and autophagy provide further mechanistic context. For example, ROS accumulation and JNK activation have been shown to act upstream of both apoptosis and autophagy in osteosarcoma cells treated with celastrol.18 Similarly, inhibition of mTOR signaling may indirectly sensitize cells to chemotherapeutic agents by disrupting survival pathways and reducing efflux pump expression.27 Future studies could examine whether these additional mechanisms contribute to the observed synergy with 5-FU. Employing three osteosarcoma cell lines with varying p53 profiles bolsters our findings, confirming that celastrol's ability to enhance chemotherapy sensitivity transcends specific genetic profiles. Still, important caveats temper the study's reach and clinical promise. Primarily, its in vitro focus, though illuminating apoptosis pathways and P-gp blockade, overlooks intricate in vivo dynamics like tumor-microenvironment crosstalk, immune responses, drug absorption, distribution, and chronic toxicity from the celastrol–5-FU pairing. Celastrol's low water solubility, quick breakdown, and slim safety margin, flaws highlighted in earlier research, could undermine real-world performance or spark unexpected side effects. Upcoming work should prioritize in vivo tests via orthotopic NOD/SCID mouse xenografts, dosing intraperitoneally to gauge tumor growth, survival rates, and tissue harm. Additionally, while P-gp suppression drives much of the synergy, we overlooked other efflux transporters like MRP1 and BCRP; probing these could reveal wider resistance-busting roles. Finally, without gain or loss-of-function assays, such as P-gp overexpression or CRISPR p53 edits, we can't firmly link these elements to the synergy's cause.
Our findings also raise important questions regarding the interplay between p53 signaling and P-gp regulation. Previous studies suggest that p53 can transcriptionally repress P-gp expression, while P-gp activity can modulate apoptotic sensitivity. The combined celastrol–5-FU treatment may therefore operate through a feed-forward loop: celastrol upregulates p53 and Bax, suppresses Bcl-2, and reduces P-gp, which in turn increases intracellular 5-FU levels and further drives apoptosis. Elucidating these interactions could provide valuable targets for future drug development. From a clinical perspective, osteosarcoma remains a challenging malignancy, with little improvement in long-term survival over the past three decades. Current regimens rely heavily on multi-agent chemotherapy, yet resistance and dose-related toxicity frequently undermine efficacy. The ability of celastrol to enhance 5-FU cytotoxicity while enabling lower drug concentrations suggests a potential to improve therapeutic outcomes while minimizing systemic side effects. Furthermore, the observed activity in p53-deficient cells is particularly relevant, as p53 mutations are common in relapsed or metastatic osteosarcoma and are often associated with chemoresistance.
Conclusion
In conclusion, this study demonstrates that celastrol potently synergizes with 5-FU to overcome chemoresistance in human osteosarcoma cells, irrespective of p53 status. Through targeted modulation of p53-dependent and -independent apoptotic pathways—evidenced by upregulated Bax and caspase-9 expression alongside downregulated Bcl-2, and inhibition of P-gp-mediated drug efflux, the combination therapy significantly enhances cytotoxicity, induces robust apoptosis, and reduces effective drug dosages. These mechanisms not only elucidate the basis for improved therapeutic efficacy but also highlight celastrol's potential as a chemosensitizer in osteosarcoma treatment, where multidrug resistance remains a formidable barrier to patient outcomes. While in vitro results are promising, further in vivo investigations and clinical trials are warranted to validate these findings and facilitate translation into effective, low-toxicity regimens for osteosarcoma management.
Research Highlights
What is the current knowledge?
-
Celastrol induces apoptosis and autophagy via ROS/JNK in osteosarcoma cells.
-
P-gp overexpression drives multidrug resistance in osteosarcoma.
-
5-FU is limited by chemoresistance in osteosarcoma treatment.
-
p53 status influences chemosensitivity and apoptosis in cancer cells.
-
Celastrol inhibits P-gp, enhancing drug retention in resistant cancer cells.
What is new here?
-
Celastrol synergizes with 5-FU, reducing IC₅₀ values 3.7- to 11.9-fold in osteosarcoma.
-
Combination therapy enhances apoptosis across p53 wild-type, null, and mutant osteosarcoma cells.
-
Celastrol downregulates P-gp by 59-72%, boosting 5-FU intracellular accumulation.
-
p53-dependent and -independent pathways mediate synergistic apoptosis in osteosarcoma.
-
Combination reduces effective drug doses, suggesting lower toxicity potential.
Competing Interests
The authors have no conflict of interest to disclose.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Ethical Approval
This study protocol was approved by the Tabriz University of Medical Sciences ethics committee, Tabriz, Iran (code: IR.TBZMED.VCR.REC.1403.351).
References
- Raymond AK, Jaffe N. Osteosarcoma multidisciplinary approach to the management from the pathologist's perspective. Cancer Treat Res 2009; 152:63-84. doi: 10.1007/978-1-4419-0284-9_4 [Crossref] [ Google Scholar]
- Ando K, Heymann MF, Stresing V, Mori K, Rédini F, Heymann D. Current therapeutic strategies and novel approaches in osteosarcoma. Cancers (Basel) 2013; 5:591-616. doi: 10.3390/cancers5020591 [Crossref] [ Google Scholar]
- Rubio-San-Simón A, Wilson W, Sironi G, le Deley MC, Palmerini E, Gaspar N. Prognostic factors in patients with relapsed high-grade osteosarcoma: a systematic review. Br J Cancer 2025; 133:1020-8. doi: 10.1038/s41416-025-03118-x [Crossref] [ Google Scholar]
- Zhra M, Akhund SA, Mohammad KS. Advancements in osteosarcoma therapy: overcoming chemotherapy resistance and exploring novel pharmacological strategies. Pharmaceuticals (Basel) 2025; 18:520. doi: 10.3390/ph18040520 [Crossref] [ Google Scholar]
- Singh U, Kokkanti RR, Patnaik S. Beyond chemotherapy: exploring 5-FU resistance and stemness in colorectal cancer. Eur J Pharmacol 2025; 991:177294. doi: 10.1016/j.ejphar.2025.177294 [Crossref] [ Google Scholar]
- Zhao LC, Tang M, Zhang QH, Hu ZY, Gao HW, Liao XY. Fabrication of a porous metal-organic framework with polar channels for 5-FU delivery and inhibiting human osteosarcoma cells. J Chem 2018; 2018:1523154. doi: 10.1155/2018/1523154 [Crossref] [ Google Scholar]
- Sun J, Wu W, Tang X, Zhang F, Ju C, Liu R. HDAC6 inhibitor WT161 performs anti-tumor effect on osteosarcoma and synergistically interacts with 5-FU. Biosci Rep 2021; 41:BSR20203905. doi: 10.1042/bsr20203905 [Crossref] [ Google Scholar]
- Sun Y, Wang C, Li X, Lu J, Wang M. Recent advances in drug delivery of celastrol for enhancing efficiency and reducing the toxicity. Front Pharmacol 2024; 15:1137289. doi: 10.3389/fphar.2024.1137289 [Crossref] [ Google Scholar]
- Zhu Y, Meng Y, Zhang J, Liu R, Shen S, Gu L. Recent trends in anti-tumor mechanisms and molecular targets of celastrol. Int J Biol Sci 2024; 20:5510-30. doi: 10.7150/ijbs.99592 [Crossref] [ Google Scholar]
- Chen Y, Ou Y, Tao Y, Liu H, Yin H, Zhong S. Effect and mechanisms of celastrol on the apoptosis of HOS osteosarcoma cells. Oncol Rep 2018; 40:2260-8. doi: 10.3892/or.2018.6619 [Crossref] [ Google Scholar]
- Yu X, Zhou X, Fu C, Wang Q, Nie T, Zou F. Celastrol induces apoptosis of human osteosarcoma cells via the mitochondrial apoptotic pathway. Oncol Rep 2015; 34:1129-36. doi: 10.3892/or.2015.4124 [Crossref] [ Google Scholar]
- Shi J, Li J, Xu Z, Chen L, Luo R, Zhang C. Celastrol: a review of useful strategies overcoming its limitation in anticancer application. Front Pharmacol 2020; 11:558741. doi: 10.3389/fphar.2020.558741 [Crossref] [ Google Scholar]
- Wang Q, Yu X, Li F, Lv X, Fu X, Gu H. Efficacy of celastrol combined with cisplatin in enhancing the apoptosis of U-2OS osteosarcoma cells via the mitochondrial and endoplasmic reticulum pathways of apoptosis. Oncol Lett 2019; 17:3305-13. doi: 10.3892/ol.2019.10007 [Crossref] [ Google Scholar]
- Lei H, Ruan Y, Ding R, Li H, Zhang X, Ji X. The role of celastrol in inflammation and diseases. Inflamm Res 2025; 74:23. doi: 10.1007/s00011-024-01983-5 [Crossref] [ Google Scholar]
- Yu X, Wang Q, Zhou X, Fu C, Cheng M, Guo R. Celastrol negatively regulates cell invasion and migration ability of human osteosarcoma via downregulation of the PI3K/Akt/NF-κB signaling pathway in vitro. Oncol Lett 2016; 12:3423-8. [ Google Scholar]
- Lei H, Ruan Y, Ding R, Li H, Zhang X, Ji X. The role of celastrol in inflammation and diseases. Inflamm Res 2025; 74:23. doi: 10.1007/s00011-024-01983-5 [Crossref] [ Google Scholar]
- Yu X, Wang Q, Zhou X, Fu C, Cheng M, Guo R. Celastrol negatively regulates cell invasion and migration ability of human osteosarcoma via downregulation of the PI3K/Akt/NF-κB signaling pathway in vitro. Oncol Lett 2016; 12:3423-8. doi: 10.3892/ol.2016.5049 [Crossref] [ Google Scholar]
- Li HY, Zhang J, Sun LL, Li BH, Gao HL, Xie T. Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: an in vitro and in vivo study. Cell Death Dis 2015; 6:e1604. doi: 10.1038/cddis.2014.543 [Crossref] [ Google Scholar]
- Moradi MT, Altememy D, Asadi-Samani M, Khosravian P, Soltani M, Hashemi L. The effect of celastrol in combination with 5-fluorouracil on proliferation and apoptosis of gastric cancer cell lines. Oncol Res 2024; 32:1231-7. doi: 10.32604/or.2024.047187 [Crossref] [ Google Scholar]
- Elshahed LF, Labah DA, Taha RM, Hassan TH, Mahmoud EF. Effect of celastrol and celastrol nanoemulsions on regeneration of mandibular bone defects in osteoporotic rats. Egypt J Histol 2025; 48:459-68. doi: 10.21608/ejh.2025.347054.2186 [Crossref] [ Google Scholar]
- Lin FZ, Wang SC, Hsi YT, Lo YS, Lin CC, Chuang YC. Celastrol induces vincristine multidrug resistance oral cancer cell apoptosis by targeting JNK1/2 signaling pathway. Phytomedicine 2019; 54:1-8. doi: 10.1016/j.phymed.2018.09.181 [Crossref] [ Google Scholar]
- Liu X, Zhao P, Wang X, Wang L, Zhu Y, Song Y. Celastrol mediates autophagy and apoptosis via the ROS/JNK and Akt/mTOR signaling pathways in glioma cells. J Exp Clin Cancer Res 2019; 38:184. doi: 10.1186/s13046-019-1173-4 [Crossref] [ Google Scholar]
- Kannaiyan R, Manu KA, Chen L, Li F, Rajendran P, Subramaniam A. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis 2011; 16:1028-41. doi: 10.1007/s10495-011-0629-6 [Crossref] [ Google Scholar]
- Tia ST, Luo M, Fan W. Mapping the role of P-gp in multidrug resistance: insights from recent structural studies. Int J Mol Sci 2025; 26:4179. doi: 10.3390/ijms26094179 [Crossref] [ Google Scholar]
- Xia YZ, Ni K, Guo C, Zhang C, Geng YD, Wang ZD. Alopecurone B reverses doxorubicin-resistant human osteosarcoma cell line by inhibiting P-glycoprotein and NF-kappa B signaling. Phytomedicine 2015; 22:344-51. doi: 10.1016/j.phymed.2014.12.011 [Crossref] [ Google Scholar]
- Li J, Duan B, Guo Y, Zhou R, Sun J, Bie B. Baicalein sensitizes hepatocellular carcinoma cells to 5-FU and epirubicin by activating apoptosis and ameliorating P-glycoprotein activity. Biomed Pharmacother 2018; 98:806-12. doi: 10.1016/j.biopha.2018.01.002 [Crossref] [ Google Scholar]
- Zhan D, Ni T, Wang H, Lv M, Sunagawa M, Liu Y. Celastrol inhibits the proliferation and decreases drug resistance of cisplatin- resistant gastric cancer SGC7901/DDP cells. Anticancer Agents Med Chem 2022; 22:270-9. doi: 10.2174/1871520621666210528144006 [Crossref] [ Google Scholar]
- Ju SM, Youn GS, Cho YS, Choi SY, Park J. Celastrol ameliorates cytokine toxicity and pro-inflammatory immune responses by suppressing NF-κB activation in RINm5F beta cells. BMB Rep 2015; 48:172-7. doi: 10.5483/bmbrep.2015.48.3.147 [Crossref] [ Google Scholar]
- Salama MM, Zaghloul RA, Khalil RM, El-Shishtawy MM. Anti-neoplastic activity of celastrol in experimentally-induced mammary adenocarcinoma in mice: targeting Wnt/β-catenin signaling pathway. NaunynSchmiedebergs Arch Pharmacol 2025; 398:14327-39. doi: 10.1007/s00210-025-04148-1 [Crossref] [ Google Scholar]
- Niu W, Wang J, Wang Q, Shen J. Celastrol loaded nanoparticles with ROS-response and ROS-inducer for the treatment of ovarian cancer. Front Chem 2020; 8:574614. doi: 10.3389/fchem.2020.574614 [Crossref] [ Google Scholar]
- Li X, Zhu G, Yao X, Wang N, Hu R, Kong Q. Celastrol induces ubiquitin-dependent degradation of mTOR in breast cancer cells. Onco Targets Ther 2018; 11:8977-85. doi: 10.2147/ott.S187315 [Crossref] [ Google Scholar]