Bioimpacts. 16:33219.
doi: 10.34172/bi.33219
Review
Checkpoint inhibition and beyond: Precision immune engineering for the immune-privileged landscape of ocular malignancies
Qamar Abuhassan Conceptualization, Methodology, Writing – original draft, 1 
Kamel Saleh Conceptualization, Project administration, Resources, Supervision, Validation, Writing – review & editing, 2, *
S. Renuka Jyothi Investigation, Methodology, Writing – original draft, 3
Radhamadhab Sahoo Formal analysis, Methodology, Writing – review & editing, 4
P. Prakash Data curation, Formal analysis, Visualization, 5
Gunjan Mukherjee Resources, Validation, Writing – review & editing, 6
Aashna Sinha Data curation, Investigation, Visualization, 7
Turabek Boyqulov Investigation, 8
Author information:
1Department of Pharmaceutics and Pharmaceutical Technology, School of Pharmacy, University of Jordan, Amman, 11942, Jordan
2Faculty of Allied Medical Sciences, Hourani Center for Applied Scientific Research, Al-Ahliyya Amman University, Amman, Jordan
3Department of Biotechnology and Genetics, School of Sciences, JAIN (Deemed to be University), Bangalore, Karnataka, India
4Department of Otorhinolaryngology (ENT), IMS and SUM Hospital, Siksha 'O' Anusandhan, Bhubaneswar, Odisha-751003, India
5Department of Biotechnology, Sathyabama Institute of Science and Technology, Chennai, Tamil Nadu, India
6University Institute of Biotechnology, Chandigarh University, Mohali, Punjab, India
7School of Applied and Life Sciences, Division of Research and Innovation, Uttaranchal University, Dehradun, Uttarakhand, India
8Department of Medicine, Termez University of Economics and Service, Termez, Uzbekistan
Abstract
Ocular malignancies, particularly uveal and conjunctival melanoma, exemplify tumors that evolve within one of the body’s most immunologically constrained ecosystems, the eye’s immune-privileged microenvironment. The limited success of PD-1/PD-L1 and CTLA-4 blockade in these cancers underscores the need to move beyond linear checkpoint inhibition toward multidimensional immune engineering. Through the confluence of synthetic bio-nanotechnology, AI-guided immunogenomics, and spatial immunomics, this review reframes ocular immunotherapy and redefines how tolerance and immunity might be programmatically regulated within ocular tissue. We synthesize recent advances in bispecific T-cell engagers, oncolytic viro-immunotherapy, mRNA and dendritic-cell vaccines, and engineered CAR/TCR-T platforms, highlighting how they collectively reconfigure the ocular tumor microenvironment from immune-silent to immune-responsive. Logic-gated antibodies, ROS-responsive nanocarriers, and CRISPR-assisted checkpoint reprogramming are added to the notion of "precision immune engineering". These developments are intended to temporarily alter immune privilege without sacrificing visual quality. Lastly, we suggest a systems-level model for ocular immuno-oncology 2.0, where immune privilege is not an unchangeable barrier but rather a configurable circuit for therapeutic orchestration. One element of a dynamic, closed-loop immune-engineering architecture is checkpoint inhibition. This platform offers the possibility of long-lasting, vision-preserving disease treatment by combining AI-driven neoantigen detection, liquid-biopsy feedback loops, and flexible delivery biomaterials. While several of these approaches remain at a conceptual or early translational stage, they outline a plausible roadmap toward vision-preserving immunotherapy in ocular oncology.
Graphical Abstract
Keywords: Ocular immuno-oncology, Checkpoint inhibition resistance, Immune-privileged tumor microenvironment, Neoantigen-driven immunotherapy, AI-guided personalized immunotherapy, Nanocarrier-based immune modulation
Copyright and License Information
© 2026 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
Nil.
Introduction
One of the human body's most immunologically specialized settings is where ocular cancers develop. Interconnected structural, immunological, and neuroimmune systems sustain this condition.1 These mechanisms directly influence how tumors respond to therapy. A defining component is anterior chamber–associated immune deviation (ACAID), whereby ocular antigen exposure promotes systemic immune tolerance rather than effector T-cell activation, favoring regulatory T-cell responses. In parallel, the blood–retinal barrier (BRB) restricts immune-cell trafficking and limits drug penetration into intraocular tissues, thereby reducing the efficacy of systemically administered immunotherapies.1 Beyond these barriers, neuroimmune modulation mediated by retinal neurons, microglia, and soluble neuropeptides actively suppresses inflammatory signaling. While this preserves visual function, it also dampens antitumor immune activity.2-5 While these mechanisms preserve visual function by limiting inflammatory damage, they also generate a microenvironment that is inherently resistant to effective antitumor immune responses.4,5
The most challenging ocular malignancy in terms of immunology and clinical management is uveal melanoma (UM). Although cutaneous melanoma and uveal melanoma originate from melanocytes, they differ substantially in their tumor ecology and immunological behavior. Uveal melanoma exhibits an extremely low tumor mutational burden, which limits neoantigen generation, restricts T-cell priming, and results in minimal pre-existing antitumor immunity.6 In addition to having a modest tumor mutation load, uveal melanoma's altered HLA expression patterns are a key immune evasion strategy.6 Rather than permanent structural abnormalities in antigen presentation, reduced HLA class I expression may develop independently of β2-microglobulin loss, indicating regulatory suppression. Simultaneously, uveal melanoma shows little increase in HLA class II expression, which is indicative of poor interferon-γ signaling and insufficient CD4 T-cell-mediated immunological support.6 Collectively, these flaws impair immune priming, reduce antigen visibility, and maintain an immunologically invisible tumor phenotype that is not very sensitive to checkpoint blockage.4,5 Antigen-presentation pathways are frequently impaired in uveal melanoma, with several studies demonstrating reduced or heterogeneous expression of HLA class I molecules on tumor cells, thereby limiting effective CD8⁺ T-cell recognition and infiltration.7 Accordingly, the classical steps of adaptive antitumor immunity, including antigen recognition, immune activation, tumor infiltration, and cytotoxic engagement, are markedly attenuated. Consequently, UM has not responded well to systemic immunotherapies that rely on pre-existing immune activation.7
These features highlight important limitations in current immunotherapeutic and immune-engineering strategies. However, UM lacks targetable surface antigens for CAR-T therapy as well as the co-stimulatory environment necessary for effective T-cell proliferation.7,8 Although TIL-based therapies can generate cytotoxic effector cells, their activity in uveal melanoma is rapidly suppressed by factors such as TGF-β and IDO. Similarly, vaccine-based approaches rarely achieve sustained intertumoral infiltration even when they induce systemic immune activation.8 In practice, systemic immune activation induced by these approaches is often rapidly neutralized by dominant metabolic and cytokine-mediated suppressive signals within the uveal melanoma microenvironment. Collectively, these limitations indicate that while existing platforms can initiate T-cell activation, they fail to maintain functional immune activity within the uveal melanoma microenvironment.8 These mechanistic limitations may also account for the disappointing outcomes seen with checkpoint inhibition alone. In contrast to UM, PD-1/PD-L1 and cytotoxic T-lymphocyte–associated antigen-4 (CTLA-4) inhibition depend on stimulating T cells that are already active in tumor identification and tumor elimination.8,9 The immune-privileged state of the eye inhibits baseline T-cell activation, and UM tumors exhibit low levels of PD-L1 and restricted interferon-response signatures. Because of this, clinical investigations typically demonstrate objective response rates of about 5–10% even with dual checkpoint blocking, and the responses that do occur are seldom long-lasting.7 These observations suggest that checkpoint inhibitors are unlikely to be effective as monotherapies and may instead require integration into broader combination strategies designed to induce inflammatory priming.8,9 All of these findings suggest that UM is a uniquely difficult immune-engineering issue, a cancer that is immunologically hidden yet pharmacologically accessible. Precision metabolic modulators, oncolytic virotherapy, T-cell redirection platforms (such as tebentafusp), and sensible combination techniques are promising methods to alter the therapeutic paradigm for this biologically resistant cancer.8,9 Together, these characteristics position uveal melanoma as a prototypical example of immune resistance driven by spatially imposed immune privilege rather than target absence, motivating a reexamination of immunotherapy from an engineering perspective.
Conceptual framework
This review is structured around a conceptual framework that links ocular immune privilege to therapeutic resistance and examines how emerging immune-engineering strategies aim to address these barriers. Accordingly, the manuscript is organized into four thematic domains: (1) the mechanisms of ocular immune privilege that produce an immune-silent tumor niche; (2) the effectiveness and limitations of existing checkpoint inhibitors within this limited ecosystem; (3) emerging precision immune-engineering strategies intended to reprogram the tumor microenvironment, such as adoptive cell technologies, TCR-based redirection, oncolytic virotherapy, and biomaterial-enabled modulation; and (4) the translational landscape of preclinical and clinical advancements that together redefine the future of immunotherapy for ocular malignancies. This structure provides a coherent link between underlying biological mechanisms, therapeutic limitations, and emerging engineering-driven solutions discussed throughout the review. Importantly, this framework does not propose a single dominant therapeutic modality, but instead emphasizes coordinated modulation of immune activation, delivery constraints, and microenvironmental resistance as the central challenge in ocular immunotherapy. This review uses a defined language framework for conceptual clarity. The phrase "precision immune engineering" refers to integrated, design-driven approaches that actively modify tumor–immune interactions by fusing biological understanding with synthetic, computational, or biomaterial-based treatments. Within this context, "immunomodulation" refers to specific, often reversible changes in the direction or strength of immunological signals, such as localized checkpoint interference, metabolic inhibition, or cytokine blocking. On the other hand, more long-lasting or systemic changes in immune activity, such as modifications to antigen presentation, T-cell differentiation stages, or immunological memory formation, are referred to as "immune reprogramming." From temporary modulation to long-term immune-state remodeling, these phrases are employed consistently throughout the article to represent varying degrees of intervention depth.
Checkpoint inhibition in ocular oncology
Checkpoint inhibition targeting CTLA-4, including agents such as ipilimumab and tremelimumab, has shown limited and variable clinical efficacy in metastatic uveal melanoma. The bulk of clinical studies have reported low objective response rates, with only occasional cases of illness stability and a lack of consistent partial or complete remissions. Although these agents are generally well tolerated, their limited effectiveness highlights the need for alternative or combination immunotherapeutic strategies tailored to the unique immunobiology of ocular malignancies. Despite a thorough clinical investigation of CTLA-4 and PD-1/PD-L1 inhibitors in uveal melanoma, a recurring pattern appears in all studies: Checkpoint inhibitor monotherapy is ineffective in uveal melanoma primarily because tumor-specific T-cell priming is profoundly impaired.6,10 Deficient dendritic-cell activation, downregulated MHC class I expression, and suppressive metabolic pathways such as IDO-mediated tryptophan depletion collectively prevent the generation and maintenance of functional effector T cells that are required for checkpoint blockade to exert therapeutic benefit.6,10 Even dual blockage produces transient responses with significant toxicity, and objective response rates seldom ever surpass 5–10%. Crucially, the underlying tumor biology of uveal melanoma, which includes its low tumor mutational burden, poor antigen presentation, lack of pre-existing T-cell priming, and dominant metabolic and cytokine-mediated immunosuppressive pathways like IDO and TGF-β signaling, is directly reflected in these modest clinical outcomes. In contrast to cutaneous melanoma, which typically exhibits high tumor mutational burden, abundant neoantigen presentation, and pre-existing T-cell infiltration, uveal melanoma lacks the immunological prerequisites required for effective checkpoint inhibition.11 Low PD-L1 expression, deficient antigen presentation, and minimal baseline interferon signaling prevent PD-1/PD-L1 and CTLA-4 blockade from restoring cytotoxic T-cell function, rendering checkpoint inhibitors largely ineffective as monotherapy in this setting.11 In this context, heterogeneity within the tumor microenvironment, rather than the intrinsic effect of checkpoint inhibition, may account for the inconsistent findings regarding the prognostic significance of liver-only metastasis.11 Collectively, existing evidence suggests that checkpoint inhibitors may be more effective when incorporated into combination regimens that induce inflammatory priming, rather than being used as stand-alone therapies.12
CTLA-4-targeting immune checkpoint blockade: medications like tremelimumab and ipilimumab
In 2011, the U.S. Food and Drug Administration (FDA) approved ipilimumab as the inaugural treatment drug for metastatic cutaneous melanoma (CM), which operates by mitigating immune suppression via the inhibition of the CTLA-4 pathway. Ipilimumab enhances cytotoxic T-cell proliferation and function by inhibiting the CTLA-4 checkpoint pathway, which normally restrains T-cell activation.13,14
Previous studies have shown that ipilimumab yields only modest therapeutic effects in patients with metastatic UM, generally leading to limited occurrences of stable disease (SD), infrequent partial responses (PR), and rare complete responses (CR), results primarily based on small-cohort research. A retrospective investigation of 20 metastatic UM patients revealed that treatment with ipilimumab resulted in a median overall survival (OS) of around five months, highlighting its limited clinical efficacy in this malignancy.15 A retrospective, population-based study assessed the therapeutic efficacy of immune checkpoint inhibitor (ICI) monotherapy versus combination regimens, seeking to ascertain if dual blockade strategies could improve clinical outcomes in patients with metastatic uveal melanoma.16 The study indicated a median OS of 9.9 months, which was comparable to, and not significantly superior to, the current benchmark median OS of approximately 10.2 months in metastatic uveal melanoma, suggesting no considerable survival benefit from the treatment method.17 In the cohort of 24 patients treated with ipilimumab, no objective responses were recorded; none attained PR or CR, thereby underscoring the limited therapeutic efficacy of CTLA-4 inhibition in metastatic uveal melanoma.16 A phase Ib/II clinical trial examining the combination of radiofrequency ablation (RFA) and ipilimumab had minimal therapeutic efficacy, since the treatment did not produce significant antitumor responses, but it was well tolerated by patients. These findings further confirm the limited clinical efficacy of ipilimumab-based strategies in metastatic uveal melanoma.15 Similar results were documented in a retrospective multicenter investigation, in which none of the 11 patients with UM attained a complete CR or PR. Only two patients (18.2%) exhibited SD, highlighting the limited responsiveness of UM to CTLA-4 inhibition.18
Tremelimumab (CP-675,206), a completely human monoclonal antibody targeting CTLA-4, was assessed in a phase II clinical trial with 11 patients diagnosed with UM. The trial indicated a median progression-free survival (PFS) of 2.9 months and a median OS of 12.8 months, reflecting a minor clinical benefit and confirming the limited efficacy of CTLA-4 inhibition in this cancer.19 A comprehensive study involving over 700 patients with UM indicated that the median OS post-diagnosis ranged from 3 to 4 months, highlighting the disease's aggressive characteristics and the absence of effective systemic therapy alternatives.20 The trial was prematurely ended at the initial interim analysis because no patients attained a complete CR or PR, indicating a lack of significant therapeutic efficacy.19 CTLA-4 inhibitors have demonstrated limited clinical efficacy in metastatic uveal melanoma, with meaningful benefit observed only in a small subset of patients. Nevertheless, these medications may provide a limited therapeutic advantage in a minor group of patients. Currently, the factors influencing individual reactions to ipilimumab are ambiguous, highlighting the necessity for biomarker-driven research to enhance the prediction and optimization of treatment results.21
Targeting the PD-1/PD-L1 axis with precision: therapeutic perspectives from atezolizumab, nivolumab, and pembrolizumab
These inhibitors act by disrupting the interaction between the programmed cell death protein-1 (PD-1) receptor and its ligand PD-L1, thereby relieving inhibitory signaling and restoring T-cell activity.22 Pembrolizumab, nivolumab, and atezolizumab are monoclonal antibodies that specifically target different elements of the PD-1/PD-L1 signaling pathway and have been granted regulatory approval for melanoma treatment. However, evaluations of their therapeutic efficacy in uveal melanoma are largely based on retrospective analyses rather than prospective trials and should therefore be interpreted with caution. Prior studies indicated that nivolumab can improve OS and PFS in patients with metastatic melanoma, chiefly by restoring cytokine release and reactivating T-cell–mediated immune responses.23 A recent single-institution trial of 14 patients with metastatic UM revealed an overall response rate (ORR) of 7.1%, suggesting that a limited number of patients saw substantial clinical improvement following PD-1/PD-L1 inhibitor therapy.24 The research indicated a median PFS of 10 weeks and a median OS of 60 weeks, with nivolumab exhibiting excellent tolerability and a tolerable safety profile during the treatment period.24 A multicenter study involving 17 patients with metastatic UM reported an ORR of 18%, with a median PFS of 5.8 months and a median OS of 10.5 months, indicating limited clinical efficacy of PD-1/PD-L1 blockade in this cohort.25 Mild to severe (grade 1–2) adverse events (AEs) such as fatigue and anorexia were the most often reported treatment-related side effects, affecting around 17% of participants. No severe (grade 3 or 4) adverse events were recorded, indicating the treatment's favorable overall tolerability.25
Concerning pembrolizumab, results from two prior expanded access programs demonstrated significant variability in ORR and PFS, while the median OS was indeterminate, underscoring the limited and inconclusive evidence regarding its clinical efficacy in metastatic uveal melanoma.26,27 A new modest, single-arm phase II trial (NCT02359851) reported a median progression-free survival of 11 months, significantly exceeding prior trials; however, the median overall survival remains uncertain.28 A prospective, single-arm cohort trial comprising 17 patients with metastatic UM documented PR in two patients (11.7%) and SD in six patients (35.3%), with a median PFS of 3.8 months and an unspecified median OS.29 Two investigations carried out by Bol et al16 and Jansen et al30—two investigations involving 43 and 9 patients with UM, respectively—exhibited similar clinical outcomes, with no instances of CR. Bol et al noted PR in three patients (7%) and SD in twelve patients (27.9%), while Jansen et al documented stable illness in five patients (56%).16,30 The median progression-free survival values were 4.8 months (about 144 days) and 18 weeks (approximately 126 days), respectively, both aligning with the data published by Rossi et al.29 The median OS values were 10.3 months (about 309 days) and 46 weeks (approximately 322 days), respectively.16,30
Previous research has assessed and contrasted the clinical outcomes linked to different anti-PD-1 and anti-PD-L1 antibodies. Algazi and colleagues31 performed an analysis of 56 patients with metastatic uveal melanoma, of which 38 were administered pembrolizumab, 16 received nivolumab, and 2 were treated with atezolizumab. The ORR was 3.6%, with a median PFS of 2.6 months and a median OS of 7.7 months. Only one patient experienced treatment termination owing to toxicity.31 A retrospective analysis indicated an ORR of 4.7% in 86 patients with metastatic UM treated with pembrolizumab or nivolumab, with median OS durations of 14 months and 10 months, respectively.32 A further retrospective investigation of 15 patients with metastatic uveal melanoma revealed no objective responses to treatment with pembrolizumab or nivolumab, indicating a median progression-free survival of 3 months and a median overall survival of 5 months.33 Recently, Koch et al18 documented an ORR of 8.9% in 45 patients with metastatic uveal melanoma treated with pembrolizumab or nivolumab. This retrospective investigation identified 11 patients who encountered AEs, with 4 patients developing severe AEs classified as grade 3 or 4.18 Indeed, more comprehensive data on PD-1 inhibitors were obtained from the IMCGp-100-202 study (NCT03070392), which encompassed the largest prospectively treated cohort to date.34 Approximately 80% of the 126 individuals in the control group underwent pembrolizumab treatment. The ORR was 5%, the disease control rate (DCR) was 27%, the median OS was 16 months, and the median PFS was 2.9 months.35 A minor subset of patients in this cohort underwent treatment with ipilimumab or dacarbazine, although the clinical outcomes showed negligible variation in comparison to the aforementioned trials.
Regarding practical practice, according to the findings of Owen et al,36 the effectiveness of future PD-1 antibody therapy was significantly affected by the time of recurrence; earlier relapses resulted in diminished treatment responses. Nevertheless, for patients who attained a positive response, the ideal length of PD-1 antibody treatment remained ambiguous. Consequently, therapy strategies necessitate additional assessment by long-term follow-up research. In general, PD-1 and PD-L1 antibodies infrequently elicited prolonged remission in patients with metastatic uveal melanoma, with merely a minor percentage demonstrating restricted responses.
Synergistic immune checkpoint blockade: Using CTLA-4 and PD-1 antibody therapy together
Due to the restricted clinical efficacy of antibody monotherapy, numerous studies have investigated the therapeutic potential of combining anti-PD-1 and anti-CTLA-4 antibodies. A phase II trial (NCT02626962) indicated an ORR of 11.5% and SD in 51.9% of 52 patients with metastatic uveal melanoma treated with a combination of nivolumab and ipilimumab.37 The median PFS was 3.0 months, while the OS was 12.7 months.37 A separate phase II trial (NCT01585194) documented an ORR of 18%, comprising one CR and five PR, in a cohort of 33 evaluable patients administered the combination of nivolumab plus ipilimumab.34 This trial revealed a median PFS of 5.5 months and a median OS of 19.1 months; nevertheless, the occurrence of serious AEs was as high as 40%.34
Multiple retrospective studies have investigated the combination of anti-PD-1 and anti-CTLA-4 antibody treatments, demonstrating similar clinical effects. A prior retrospective research indicated a median PFS of 2.8 months in 15 patients with metastatic uveal melanoma (12 of whom were assessable) using a PD-1 inhibitor in conjunction with ipilimumab.32 Two supplementary investigations revealed analogous results: one included 64 patients with metastatic UM treated with nivolumab or pembrolizumab in conjunction with ipilimumab, while the other had 89 metastatic UM patients administered the nivolumab–ipilimumab regimen.38,39 The initial research recorded a median PFS of 3 months and a median OS of 16.1 months.38 Simultaneously, 39.1% of the patients encountered severe AEs, with 37.5% categorized as grade 3 and 1.6% as grade 4.38 The aforementioned study indicated an ORR of 11.6%, a median PFS of 2.7 months, a median OS of 15 months, and a 30% occurrence of severe AEs.39 A retrospective, population-based study encompassed 19 patients who underwent combination therapy with ipilimumab and nivolumab.16 The median PFS was 3.7 months, while the OS was 18.9 months.16 A retrospective case series of eight patients with metastatic uveal melanoma assessed the therapeutic efficacy of combining ipilimumab and nivolumab with trans arterial chemoembolization.40 The median OS was 14 months; however, the median PFS was not disclosed.40
The largest retrospective multicenter study to date stratified 178 patients with metastatic uveal melanoma into two cohorts: cohort A, comprising 55 patients with liver-only metastases, and cohort B, comprising 123 patients with both hepatic and extrahepatic metastases.18 Ninety-four patients (34 from cohort A and 60 from cohort B) received combination therapy with anti-PD-1 and anti-CTLA-4 drugs. Furthermore, 31.2% of the patients encountered severe AEs, with no significant disparity noted across cohorts A and B.18 The median PFS for the total cohort was 2.8 months (2.4 months in cohort A compared to 2.9 months in cohort B), while the median OS was 16 months (6.1 months in cohort A versus 18.2 months in cohort B).18 While the median progression-free survival was similar between the two groups, cohort B demonstrated a longer median overall survival than cohort A. Patients with both hepatic and extrahepatic metastases exhibited a more favorable response to dual immune checkpoint blockade (ICB) therapy and attained enhanced survival outcomes relative to those with liver-only metastases.18 The mechanism underlying this therapeutic action is yet unclear and requires further exploration. Concerning treatment-related adverse events (TRAEs) and treatment-related serious adverse events (TRSAEs), both phase II trials had analogous toxicity profiles. Almost all patients encountered TRAEs, including diarrhea or colitis, tiredness, dermatological reactions, hepatic incidents, and hypothyroidism. TRSAEs were noted in almost 50% of the patients, predominantly manifesting as diarrhea, hepatic problems, and fever.34,37 Fatalities associated with treatment were infrequent, with documented instances of thyroiditis and Guillain–Barré syndrome.37 Nonetheless, the toxicity profile is moderate and shows minimal variation from that seen in CM, indicating its promise as a feasible therapeutic strategy.37 The clinical results of metastatic uveal melanoma are much inferior to those of metastatic cutaneous melanoma. A contributing cause is the much-reduced mutational burden in both primary and metastatic uveal melanoma, averaging 0.5 mutations per megabase, in contrast to 49.2 mutations per megabase in cutaneous melanoma.41,42 The decreased mutation burden may lead to attenuated immune activation and lower neoantigen production.43 A further contributing element is the markedly reduced expression of PD-1 and PD-L1 in UM metastases relative to CM metastases.44 Additionally, lymphocyte-activation gene 3 (LAG-3) has been recognized as the primary marker of exhaustion, potentially elucidating the restricted effectiveness of CTLA-4 and PD-1/PD-L1 inhibitors.45
In summary, most patients who responded to ICI medication attained only PR, and the duration of therapeutic improvement was constrained. As of now, neither the National Comprehensive Cancer Network (NCCN) nor the American Society of Clinical Oncology (ASCO) has included immune checkpoint inhibitors (ICIs) in their treatment guidelines, despite the prior FDA approval of nivolumab and pembrolizumab as adjuvant therapies for melanoma patients with lymph node involvement after complete tumor resection.46,47 Additional high-quality randomized controlled trials are necessary to further validate this therapeutic approach, due to the current deficiency of prospective, large-sample, evidence-based data. Nevertheless, this regimen, especially the combination of anti-PD-1 and anti-CTLA-4 therapy, continues to represent a potentially advantageous choice for patients with otherwise restricted therapeutic possibilities.
Targeting possible immune checkpoints
Novel immunological checkpoints such as TIGIT have demonstrated potential as innovative immunotherapeutic targets in UM. Preclinical studies demonstrate that TIGIT is increased in malignancies and that its concurrent inhibition with PD-1 yields synergistic effects by augmenting T-cell activation. Although monoclonal antibodies targeting TIGIT are presently in clinical trials for numerous malignancies, their therapeutic efficacy in uveal melanoma needs to be thoroughly clarified.
Therapeutic advances using T-cell immunoreceptor ITIM/ITSM domain inhibitors to target TIGIT checkpoint signaling
The receptor contains the immunoreceptor tyrosine-based inhibitory motif (ITIM) domain. TIGIT is an inhibitory molecule found on lymphocytes that diminishes the function of T cells and natural killer (NK) cells by interacting with CD155 on antigen-presenting cells (APCs) or tumor cells.48 In the study conducted by Chauvin et al49 an increase in TIGIT expression, accompanied by co-expression of PD-1, was noted in melanoma patients. Furthermore, TIGIT expression was seen to escalate subsequent to PD-1 blockage. The simultaneous suppression of TIGIT and PD-1 receptors resulted in increased degranulation and proliferation of CD8⁺ T cells when exposed to cells that express the TIGIT ligand.49 Recently, in the study conducted by Stalhammar et al50 primary UM tumors demonstrate a markedly elevated mean density of TIGIT-positive cells per mm2 in comparison to normal choroidal tissue, paralleling the disparity noted between metastatic lesions and normal liver tissue. Furthermore, metastatic primary uveal melanoma exhibits a higher density of TIGIT-positive cells per mm2 compared to both non-metastatic and corresponding metastatic uveal melanoma samples. The findings indicate that TIGIT is a promising target for immunotherapy in UM. Multiple monoclonal antibodies targeting TIGIT, including iragolumab, AB-154, BMS-986207, and MK-7684, have been produced.51 Clinical trials assessing TIGIT inhibition have commenced for numerous cancer types, including multiple myeloma and chronic myeloid leukemia. Nonetheless, its therapeutic efficacy in advanced UM is yet to be established and necessitates further exploration.
IDO blockage
Indoleamine 2,3-dioxygenase (IDO) is a rate-limiting metabolic enzyme that facilitates the conversion of tryptophan, consequently affecting the proliferation, activation, and survival of lymphocytes.52,53 Research indicates that IDO inhibits the function of T cells and NK cells while facilitating tumor angiogenesis.53 In UM cells, the elevation of IDO expression generated by interferon-gamma (IFN-γ) can shield tumor cells from T-cell and NK-cell-mediated immune responses, thus promoting immune evasion.53 Notably, the combination of IDO1 inhibitors with other therapeutic drugs frequently produces superior clinical outcomes compared to IDO1 inhibitor monotherapy, due to their synergistic effects.52 A phase I/II clinical trial (ECHO-202/KEYNOTE-037) demonstrated encouraging antitumor activity and favorable tolerability for the combined therapy of the IDO1 inhibitor epacadostat and pembrolizumab; however, patients with UM were excluded from this research.54 Results from a phase III clinical trial (NCT02752074) demonstrated that this medication did not enhance PFS or OS in patients with unresectable or metastatic melanoma.55 In the study led by Stalhammar et al,50 both metastatic and nonmetastatic main tumors demonstrated a greater mean density of IDO-positive cells per mm2 compared to normal choroidal tissue, a trend similarly noted in metastases when compared to normal liver tissue. Furthermore, IDO expression was associated with the expression of the checkpoint receptor TIGIT, and both exhibited a moderate connection with the immune-related prognostic signature.50,56 IDO may serve as a promising immune checkpoint target for UM; nevertheless, despite the research on many IDO-targeting drugs, none of the current studies involve patients with UM.
LAG3
LAG3 is a receptor present on NK cells, T cells, and plasmacytoid dendritic cells, newly recognized as an immunological checkpoint protein.57,58 LAG-3 signaling in T cells may induce T-cell malfunction, facilitating tumor immune evasion.58 In the study conducted by Woo et al,59 LAG-3 and PD-1 were shown to be co-expressed on tumor-infiltrating lymphocytes (TILs), functioning synergistically to augment T-cell proportions and preserve immunological homeostasis. The simultaneous inhibition of these receptors demonstrated a reduction in tumor proliferation and an augmentation of anticancer immune responses.59 In melanoma patients who exhibited disease progression following previous anti-PD-1/PD-L1 therapy, the combination of the anti-LAG3 antibody BMS-986016 with nivolumab showed clinical efficacy. Regrettably, there is presently no clinical evidence substantiating the effectiveness of anti-LAG3 therapy in UM. Single-cell study of UM demonstrated that, among exhaustion-associated immune checkpoint markers on CD8⁺ T cells, LAG3 displayed the highest expression level, whereas PD-1 exhibited the lowest.45 Furthermore, the expression of LAG3 and its ligand Galectin-3 exhibited a strong correlation with high-risk clinical and histological features, including the epithelioid cell type, absence of BAP1 expression, and monosomy of chromosome 3.58 These findings suggest that LAG-3 represents a potentially relevant immune checkpoint target in uveal melanoma, warranting further clinical investigation (Table 1).58 All of these clinical results highlight the importance of immune checkpoint inhibition as a clinical baseline in uveal melanoma, but they also highlight how ineffective it is as a stand-alone tactic in the immune-privileged ocular microenvironment. As a result, immune-engineering techniques that actively promote priming, trafficking, and persistence are required.
Table 1.
Clinical outcomes and prospective avenues for immune checkpoint inhibition in uveal melanoma
|
Checkpoint axis
|
Key agents
|
Clinical signal
|
Challenges
|
Future perspectives
|
References
|
| CTLA-4 |
Ipilimumab and Tremelimumab |
Mostly SD; rare PR/CR; OS ~5–12 mo |
Low T-cell priming, weak immunogenicity |
Dual blockade; biomarker-driven use; liver-directed combos |
60
|
| PD-1/PD-L1 |
Nivolumab, Pembrolizumab, Atezolizumab |
ORR 3–18%; PFS 2–5 mo; OS 7–16 mo |
Low TMB, weak PD-L1, early relapse |
Precision-guided use; combos with vaccines/BiTEs |
61
|
| Dual ICB |
Nivolumab + Ipilimumab |
ORR 11–18%; PFS ≤5.5 mo; OS ≤19 mo; AEs 30–40% |
High toxicity; variable benefit by metastasis site |
Dose de-escalation; triplet blockade (LAG-3/TIGIT) |
62
|
| TIGIT |
Iragolumab, AB-154, MK-7684 |
No UM trials; upregulated in UM; PD-1 co-expressed |
Functional redundancy; untested in UM |
First trials in TIGIT+ UM; combo with PD-1 |
63
|
| IDO |
Epacadostat, Linrodostat |
No UM data; melanoma trials failed |
Promotes immune escape & angiogenesis; TIGIT-linked |
Dual metabolic–immune inhibition; metabolic biomarkers |
64
|
| LAG-3 |
Relatlimab, Favezelimab |
No UM data; highest CD8+ exhaustion marker |
Ligand (Galectin-3) linked to high-risk UM |
Anti-LAG3 + PD-1 in biomarker-enriched cohorts |
65
|
Immunotherapy redefined beyond checkpoint blockade
Beyond immune checkpoint blockade, several emerging immunotherapeutic strategies have been investigated to address resistance mechanisms and improve treatment efficacy in uveal melanoma. Strategies such as cancer vaccinations, adoptive cell transfer, and metabolic regulation are expanding the treatment landscape. Together, these methods are not intended to serve as checkpoint inhibition's direct clinical substitutes, but rather as translational platforms intended to get around particular biological bottlenecks found in uveal melanoma and to guide logical combination strategies for further clinical development. Unlike systemic tumors, uveal melanoma thrives within the immune-privileged ocular niche, where complex inhibitory networks suppress both innate and adaptive immune responses. Fig. 1 schematically summarizes the multidimensional immune-escape mechanisms that inform the development of advanced immunotherapeutic strategies.
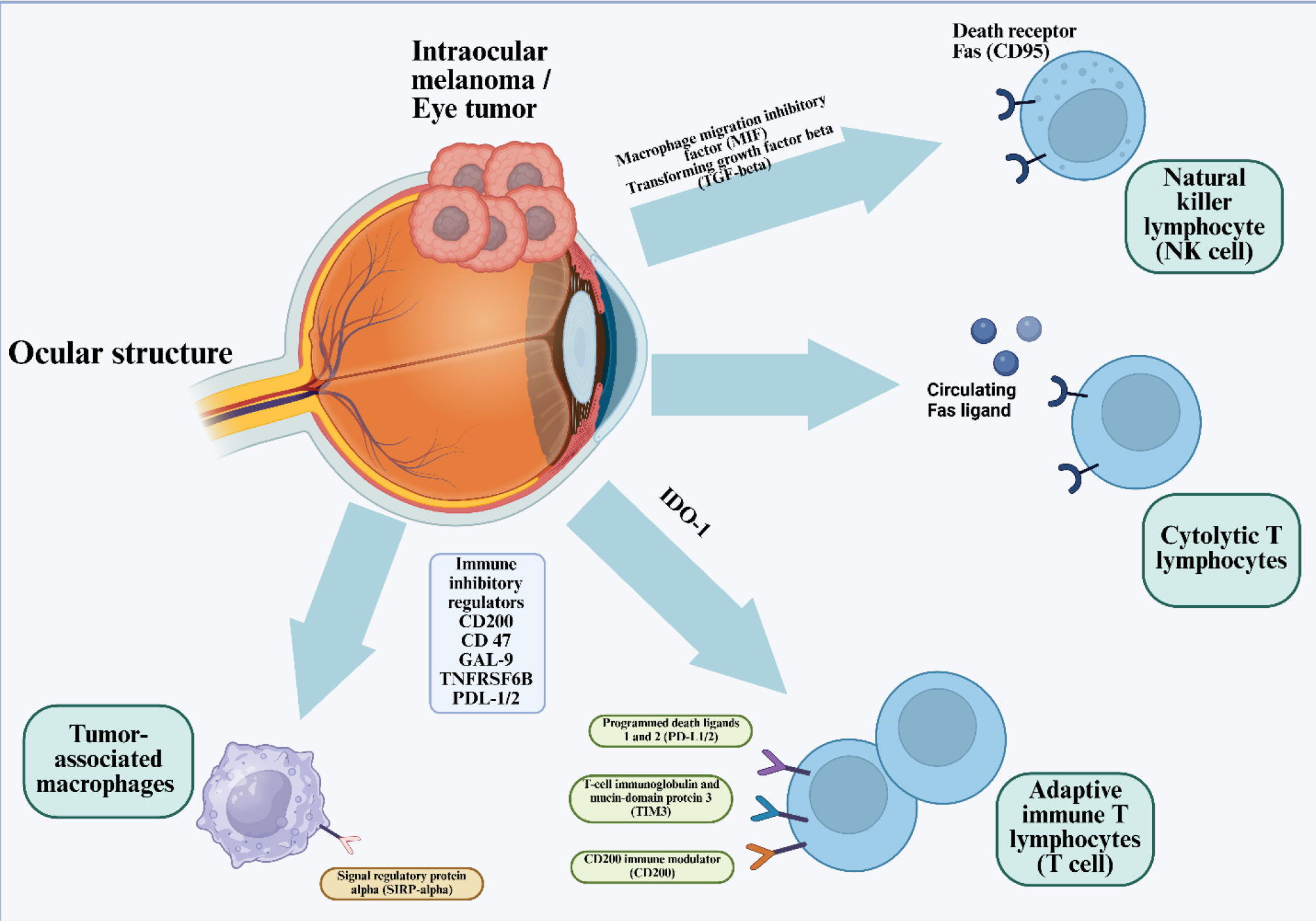
Fig. 1.
Immune-Escape Architecture of Uveal Melanoma within the Immune-Privileged Ocular Microenvironment. This schematic illustrates the multidimensional immunosuppressive landscape that enables uveal melanoma to persist within the immune-privileged environment of the eye. Tumor cells release macrophage migration inhibitory factor (MIF) and transforming growth factor-β (TGF-β), establishing a tolerogenic milieu that dampens both innate and adaptive cytotoxic responses. Expression of indoleamine-2,3-dioxygenase-1 (IDO-1) further depletes tryptophan, metabolically arresting T-cell proliferation. Tumor-derived immune inhibitory regulators including CD200, CD47, Galectin-9 (GAL-9), TNFRSF6B (DcR3), andPD-L1/2 collectively suppress effector immune infiltration and activation. On the effector side, NK lymphocytes expressing death receptor Fas (CD95)become inactivated by tumor-secreted circulating Fas ligand (FasL), whilecytolytic T lymphocytesencounter apoptotic blockade through similar Fas–FasL signaling. Adaptive immune T cells exhibit overexpression of multiple inhibitory receptors,PD-1, TIM-3, and CD200R, which attenuate cytotoxic potential and sustain T-cell exhaustion. Simultaneously, tumor-associated macrophages (TAMs)expressingsignal regulatory protein-alpha (SIRP-α)interact with tumor CD47 to deliver the “don’t-eat-me” signal, preventing phagocytosis and maintaining immune evasion. Together, these interlocking pathways define a self-reinforcing network of immune privilege that shields ocular tumors from systemic immune clearance.43 Adapted from Basile et al.43 (distributed under the terms of the Creative Commons Attribution License (CC BY).
.
Immune-Escape Architecture of Uveal Melanoma within the Immune-Privileged Ocular Microenvironment. This schematic illustrates the multidimensional immunosuppressive landscape that enables uveal melanoma to persist within the immune-privileged environment of the eye. Tumor cells release macrophage migration inhibitory factor (MIF) and transforming growth factor-β (TGF-β), establishing a tolerogenic milieu that dampens both innate and adaptive cytotoxic responses. Expression of indoleamine-2,3-dioxygenase-1 (IDO-1) further depletes tryptophan, metabolically arresting T-cell proliferation. Tumor-derived immune inhibitory regulators including CD200, CD47, Galectin-9 (GAL-9), TNFRSF6B (DcR3), andPD-L1/2 collectively suppress effector immune infiltration and activation. On the effector side, NK lymphocytes expressing death receptor Fas (CD95)become inactivated by tumor-secreted circulating Fas ligand (FasL), whilecytolytic T lymphocytesencounter apoptotic blockade through similar Fas–FasL signaling. Adaptive immune T cells exhibit overexpression of multiple inhibitory receptors,PD-1, TIM-3, and CD200R, which attenuate cytotoxic potential and sustain T-cell exhaustion. Simultaneously, tumor-associated macrophages (TAMs)expressingsignal regulatory protein-alpha (SIRP-α)interact with tumor CD47 to deliver the “don’t-eat-me” signal, preventing phagocytosis and maintaining immune evasion. Together, these interlocking pathways define a self-reinforcing network of immune privilege that shields ocular tumors from systemic immune clearance.43 Adapted from Basile et al.43 (distributed under the terms of the Creative Commons Attribution License (CC BY).
Cancer vaccines
Cancer vaccines employ tumor cells or modified antigens to induce CD4⁺ and CD8⁺ T-cell responses. Although preclinical studies have yielded encouraging findings, clinical outcomes in uveal melanoma remain modest and largely anecdotal.
Techniques for tumor-derived whole-cell vaccination
The premise of whole cell-based vaccinations is that tumor cells can act as non-professional APCs and are notably able to produce unique tumor antigen peptides via unconventional pathways that significantly differ from those employed by professional APCs.66 Verbik et al attributed the suppression of CD8⁺ T-cell activation in UM cells to the absence of HLA class II expression or co-stimulatory molecules. Consequently, through genetic manipulation, UM cells can be designed to express recipient syngeneic MHC II (HLA II or HLA-DR) alleles in conjunction with CD80 co-stimulatory molecules.67 MHC II UM vaccines are produced from genetically engineered UM cells. Specifically, CD80 molecules can mitigate the IFN-γ-induced elevation of PD-L1, thus reducing T-cell suppression. Simultaneously, the inserted MHC II molecules, lacking the MHC II-associated invariant chain (Ii), can bind atypical tumor peptides and facilitate antigen presentation via unusual intracellular trafficking routes.66 Significantly, although matching of MHC I alleles is not obligatory, MHC II molecules must possess at least one compatible allele with the patient in instances of UM.51
The primary objective of vaccination is to stimulate CD4⁺ T cells. These cells are essential for supporting CD8⁺ T-cell–mediated immunity and the development of durable immunological memory.68 Initially, they operate as conventional “helper” T cells, releasing diverse cytokines crucial for the activation and functionality of CD8⁺ T cells.69,70 Secondly, they induce dendritic cells (DCs) to express CD40 molecules, a process termed “licensing” which subsequently activates CD8⁺ T cells.71,72 Additionally, CD4⁺ T lymphocytes can directly promote cytotoxicity in tumor cells via mechanisms including Fas-mediated cytolysis or apoptosis triggered by tumor necrosis factor–related apoptosis-inducing ligand (TRAIL).73,74 Vaccine-induced CD4⁺ T cells can identify and react to initial UM cells, while also exhibiting cross-reactivity with metastatic UM cells. Similarly, activated CD8⁺ T lymphocytes demonstrate cytolytic efficacy against both primary and metastatic uveal melanoma cells.66,68 In a recent study, Kittler et al75 offered a comprehensive elucidation of the Mel202/DR1/CD80 vaccines, which proficiently primed and activated highly pure CD4⁺ T cells. The activated CD4⁺ T cells subsequently proliferated, produced IFN-γ, and formed a varied polyclonal CD4⁺ T-cell repertoire, including T helper (Th) type 1, Th2, Th17, and regulatory T (Treg) cells.75 Among these subgroups, Treg cells seem to exert no considerable influence on the overall efficacy of the anticancer vaccination response.75 Currently, clinical data are limited to exceptionally rare or anecdotal instances with positive outcomes.
Dendritic cell-based vaccination strategies that present antigens
DCs, with the highest antigen-presenting efficacy, are the sole professional APCs capable of activating naïve antigen-specific T lymphocytes. Consequently, employing dendritic cells to provoke immunologic anticancer responses and formulate dendritic cell-based vaccines constitutes a logical approach for managing stage IV melanoma.76 This method involves transfecting autologous monocyte-derived DCs with particular mRNA sequences that encode target antigens to generate optimal DC vaccines. The tumor antigens gp100 and tyrosinase are predominantly expressed in UM cells, making them optimal targets for UM immunotherapy.77,78 In a Phase II research, Bol et al79 DCs were transfected with mRNA expressing two antigens to present HLA-A*02:01-restricted peptides, thereby producing DC vaccines that can elicit and amplify tumor-specific CD8⁺ and CD4⁺ T-cell responses. The median disease-free survival was 34.5 months, and the 3-year OS rate of 79% in DC-vaccinated patients surpassed the normal literature figure of approximately 60% for high-risk uveal melanoma. Moreover, researchers proposed that DC immunization may induce de novo immune responses and demonstrated little harm when utilized as an adjuvant therapy in high-risk UM patients.79 Currently, there is no clinical evidence indicating that dendritic cell (DC) vaccine immunotherapy is more effective than other immune-based or non-immune therapeutic modalities.17,80
A current randomized phase III clinical trial (NCT01983748) aims to evaluate the efficacy of DC vaccines containing autologous tumor RNA in preventing or delaying disease development in high-risk UM patients, in comparison to standard therapy. A phase I trial (NCT04335890) is now examining the safety, tolerability, and possible overall survival benefit of IKKβ-matured, RNA-loaded dendritic cell vaccines in patients with metastatic uveal melanoma.
Adoptive cell treatments
Cell transfer by adoption
Adoptive cell transfer (ACT) involves the ex vivo expansion and activation of autologous immune cells followed by their reinfusion into the patient. This tailored immunotherapy generally employs CD8⁺ T cells or tumor-specific CD8⁺ T cells, although CD4⁺ helper (Th) cells may also be utilized. These cells are frequently sourced from TILs acquired from biopsies of UM patients and may be genetically modified to identify particular tumor antigens. Strobel et al have conducted a comprehensive assessment of this therapeutic approach; hence, it is only succinctly reviewed in this chapter, accompanied by updates on newly commenced clinical studies.
The regression of UM produced by the adoptive transfer of autologous TIL was initially reported by Chandran et al.81,82 Recently, numerous studies have been investigating innovative ACT-based methodologies. BPX-701 is a T-cell product wherein autologous T cells are transduced with an HLA-A2-restricted, PRAME-specific T-cell receptor (TCR) and integrated with an inducible caspase-9 safety switch (NCT02743611). PRAME, a melanoma antigen preferentially expressed, has been identified in around fifty percent of both primary and metastatic uveal melanoma cases.83,84 An alternative strategy utilizes autologous CD8⁺ T lymphocytes targeting SLC45A2, an antigen present in 100% of UM cell lines, although expressed at minimal or undetectable levels in normal tissues (NCT03068624). These T cells, easily produced from donor sources, can effectively eradicate most HLA-matched melanoma cells.85 The third method utilizes MAGE-C2/HLA-A2 TCR T cells (NCT04729543). MAGE-C2, a constituent of the cancer germline antigen gene subfamily, is solely expressed in neoplastic tissues. The antigenic peptide originating from MAGE-C2 can elicit specific T-cell responses in select patients without inducing measurable harm.86,87 Nevertheless, none of this research disclosed the findings.
CAR-T cell immunotherapy with engineering
Chimeric antigen receptor (CAR) T-cell therapy is a novel immunotherapeutic approach that has demonstrated efficacy in treating several hematologic malignancies in recent years. This method entails the incorporation of genetic alterations into enriched T cells, generally with retroviral vectors like lentiviruses or employing precise gene-editing techniques, to facilitate the expression of CARs on the T-cell surface.88 The modified T-cells are subsequently grown to a level suitable for therapeutic application.88 Chimeric Antigen Receptors (CARs) are designed cell surface receptors that generally consist of a target-binding extracellular domain, a hinge region, a transmembrane domain that secures the receptor to the cell membrane, and one or more intracellular signaling domains that facilitate T-cell activation.89 By integrating target-binding single-chain variable fragments (scFv) with specific intact surface antigens, CAR T cells can identify targets irrespective of HLA molecular context, rendering them widely applicable among patients with varied HLA backgrounds.89,90
In the study conducted by Forsberg et al,91 it was demonstrated that, among the established CAR-T targets, HER2 mRNA was the sole molecule expressed at a considerable level in the majority of UM samples, and that UM cells could respond to HER2 CAR-T cells in a target-specific way. HER2 CAR-T cells successfully eliminated UM cells in both in vitro conditions and in human IL-2 transgenic NOD/SCID IL-2 receptor gamma-knockout mice.91 This treatment elicited significant or total regression of UM refractory to conventional ACT therapy using autologous TILs, indicating a promising novel therapeutic approach and facilitating the clinical application of CAR-T technology. While the anticancer efficacy of HER2 CAR-T cells has been established in both in vivo and in vitro models, additional studies are necessary to assess their therapeutic applicability and long-term outcomes in UM patients.91 A phase I clinical trial (NCT03635632) is now examining C7R-GD2 CAR-T cells for the treatment of various GD2-positive solid tumors, including GD2-positive uveal melanoma. This study seeks to assess the tolerability, safety, and therapeutic effectiveness of these modified CAR-T cells. The C7R gene was integrated into GD2 CAR-T cells to ensure prolonged cytokine signaling and augment their persistence, therefore enhancing overall survival and functional longevity. Furthermore, RNA-interference strategies targeting metabolic and oxidative-stress regulators, such as siRNA-mediated silencing of PFKFB4 and HMOX1, have been shown to suppress tumor proliferation and enhance sensitivity to chemotherapeutic and immune-based regimens.92 The research reveals a significant molecular contradiction across vaccination, TIL, CAR-T, and TCR-T modalities: cellular treatments seldom ever produce long-lasting responses, even though UM expresses highly targetable antigens such as gp100, PRAME, and SLC45A2.93 This suggests that effector cells' incapacity to operate in the immune-privileged and metabolically restrictive milieu is the main obstacle rather than antigen shortage. DC vaccines demonstrate a separation between immune activation and successful tumor penetration by producing substantial systemic immunogenicity but little tumor control.10 In rare instances, TIL and modified TCR-T cells produce striking regressions; nonetheless, they often die from TGF-β, IDO-mediated inhibition, and quick fatigue. The absence of UM-specific surface antigens presents further difficulties for CAR-T treatments. Together, our results demonstrate that before cell-based treatments may be clinically transformational in UM, they need microenvironment-modifying primers, such as metabolic inhibitors or oncolytic viruses.94
Translational constraints and risk–benefit considerations of adoptive cell therapies in ocular tumors
Although adoptive cell-based treatments, such as CAR-T and TCR-engineered lymphocytes, present an intuitively appealing approach to antigen-directed tumor removal, using them to treat ocular cancers presents a number of biological and safety issues that are very different from those faced by cutaneous melanoma or hematologic cancers.94 One major limitation concerns target specificity. Many candidate antigens in uveal melanoma are not strictly tumor-exclusive, increasing the risk of on-target/off-tumor toxicity,93 a worry that is especially important for an organ where even little collateral damage might cause permanent blindness. Ineffective lymphocyte trafficking and persistence constitute a significant obstacle in addition to antigen selection.95 Effective infiltration and long-term activity of transplanted effector cells are limited by the immune-privileged ocular milieu, vascular-stromal limitations, and suppressive soluble mediators (such as TGF-β and IDO).96 Rapid depletion and limited access to ophthalmic and hepatic metastatic sites often reduce the in vivo performance of systemically expanded cytotoxic lymphocytes, even when they exhibit strong activity ex vivo.97 Cytokine-associated toxicity is another factor to take into account. In ocular oncology, where therapeutic benefit must be weighed against stringent safety thresholds and maintenance of neurologic and visual function,97 cytokine release syndrome and immune effector cell–associated neurotoxicity, both of which have been extensively described in systemic CAR-T applications, present heightened concerns.98 These hazards limit the therapeutic window for cell-based therapies and restrict the flexibility of dosage.98 Lastly, adaptive immune evasion and antigen heterogeneity continue to be major challenges. Antigen expression in uveal melanoma varies both spatially and temporally, allowing antigen-low or antigen-negative clones to proliferate selectively under immunological challenge.99 This phenomenon highlights the necessity for multispecific or combinatorial techniques and restricts the durability of responses when single-target tactics are used.97-99 When taken as a whole, these factors indicate that although adoptive cell therapies have conceptual potential, their clinical impact in ocular tumors will probably depend on improved antigen selection, the inclusion of intrinsic safety controls, tactics to improve trafficking and persistence, and logical integration with therapies that modify the microenvironment rather than being used as stand-alone modalities.96-99
Virotherapy with oncolytics
Clinical advancement
The notion of employing viruses for cancer therapy originated in the 1950s, paralleling advancements in tissue culture techniques and rat cancer models.100 Throughout that period, numerous cancer patients received treatment with raw oncolytic viral formulations, including contaminated bodily fluids, administered via practically all possible routes.101 Although the immune system typically neutralized the viruses before they could influence tumor progression, occasional infections succeeded, leading to tumor regression, particularly in immunocompromised patients, who, however, often became ill or died once the infection spread to healthy tissues. A notable example came from Osaka University, where tumor regression was observed in 37 out of 90 terminal cancer patients treated with a wild-type mumps virus. Despite the encouraging outcomes, this research was discontinued in the 1970s, and the viral strains used were eventually lost (T. Asada, personal communication). The advent of modern oncolytic virotherapy, where viral genomes are genetically modified to enhance tumor selectivity, can be traced to a 1991 study demonstrating that a thymidine kinase (TK)-deficient herpes simplex virus (HSV) with reduced neurovirulence exhibited anti-tumor activity in a murine glioblastoma model.102 Since the inception of viral engineering for the creation of an oncolytic HSV, there has been swift clinical advancement in this domain. Numerous completed and ongoing trials now utilize oncolytic viruses from at least ten different viral families, with a steady introduction of newly developed oncolytic viruses entering clinical evaluation and application.103,104
Oncolytic viruses have generally demonstrated acceptable tolerability across a range of clinical studies.105 Nonetheless, forthcoming clinical trials utilizing oncolytic viruses are anticipated to utilize elevated dosages, as continuous technological innovations such as enhanced cell substrate optimization, the integration of cell microcarriers, and the utilization of disposable wave bioreactors persist in improving manufacturing efficiency and viral production yields.106-108 Consequently, it may still be premature to ascertain if successful oncolytic virotherapy may be attained without considerable toxicities at therapeutically beneficial levels. A specific safety worry pertains to the potential for an oncolytic virus to disseminate outside the treated patient and perhaps undergo mutation, so reacquiring its original harmful characteristics.109 Nonetheless, despite sporadic reports of viral shedding in bodily fluids like urine or respiratory secretions, there is no recorded evidence of oncolytic virus transmission to caregivers or close contacts.105
Clinical efficacy
Recent phase 1/2 clinical trials have provided evidence for the efficacy of single-agent oncolytic virotherapy, further substantiated by compelling anecdotal case reports.100,110-112 In a clinical experiment, talimogene laherparepvec, formerly referred to as OncoVEX, an oncolytic HSV designed to express granulocyte-macrophage colony-stimulating factor (GM-CSF), was directly administered into the tumors of patients with metastatic malignant melanoma. The treatment led to total remission of both injected and uninjected lesions in 8 of the 50 individuals who underwent therapy.110 This study is the most compelling evidence that intratumoral administration of an oncolytic virus can successfully cross-prime and augment anticancer immune responses. Melanoma, due to its recognized sensitivity to immunotherapy, is particularly amenable to oncolytic virotherapy, with positive results with both HSV-GM-CSF (OncoVEX) and vaccinia virus-based therapies.113
In the second clinical trial, JX594, an oncolytic vaccinia virus genetically engineered to express GM-CSF, was administered through intertumoral injection to patients with unresectable hepatocellular carcinoma, yielding objective tumor responses in 3 of the 10 evaluable subjects.111 Clinical trials integrating oncolytic viruses, including reovirus, vaccinia, and HSV, with pharmacological agents or radiation are yielding a significant incidence of tumor responses.111,114-116 Nonetheless, it is unclear whether the documented therapeutic responses are directly due to the oncolytic viruses or the simultaneous administration of potent anticancer agents in combination therapy. This crucial question can only be definitively resolved by randomized phase 3 clinical studies. To date, clinical evidence has not clearly established direct viral oncolysis as the dominant mechanism of tumor elimination.105 Consequently, the fundamental principle of the oncolytic paradigm, where a systemically administered virus extensively spreads across tumor locations to provoke tumor eradication, remains to be definitively substantiated. A recent phase I clinical trial indicated that JX594, delivered intravenously, was detectable in tumor biopsy samples only when the circulating viral burden surpassed a threshold of 10⁹ infectious units.117 This study's primary conclusion is that systemically administered oncolytic viruses can specifically target tumor locations by traversing tumor-associated blood arteries and proliferating within the malignant tissue. This process is reliant on concentration and becomes observable only when the viral dose exceeds a crucial threshold. Thus, the degree of direct tumor ablation caused by oncolytic activity seems to be significantly dosage-dependent, while the attainable dose for numerous oncolytic viruses is often restricted by manufacturing constraints. Additional insights have arisen from both active and concluded clinical trials. Initially, as clinical outcomes often do not meet initial expectations, it is clear that more predictive and physiologically appropriate preclinical models are necessary. There is an urgent requirement for orthotopic cancer models in immunocompetent animals that are both vulnerable to the specific oncolytic virus under investigation and accurately mimic the pathogenesis of human infection. Current models frequently prove insufficient due to either the absence of a functional immune system, as observed in cultured cell lines or human xenograft models, or their inherent resistance to the virus being studied, with certain exceptions like the vaccinia virus. An additional insight is that iterative phase I clinical trials may become standard practice in oncolytic virotherapy. In contrast to traditional pharmaceuticals, which are usually optimized prior to clinical trials, oncolytic viruses function as intricate engineering systems with multiple adjustable components that can be perpetually adjusted and enhanced. Iterative phase I studies offer a pragmatic framework for integrating incremental genetic and design improvements into the clinical development process without obstructing regulatory or safety protocols. A significant advancement from recent clinical research is the ability to monitor viral spread in patients via the production of reporter transgenes, providing essential pharmacokinetic data throughout early development. In rat research, viral dissemination can be assessed post-mortem by examining the distribution of infected cells at various time intervals; however, such invasive methods are impractical in human trials. This constraint has impeded comprehension of why oncolytic virotherapy results in humans frequently falling short compared to preclinical models. To address this, reporter genes have been integrated into oncolytic viral genomes, facilitating recurrent, noninvasive imaging and quantification of viral infection dynamics throughout the body.118,119
Administration of an oncolytic measles virus engineered to express the soluble extracellular domain of carcinoembryonic antigen (MV-CEA) via intraperitoneal injection in patients with treatment-resistant ovarian cancer revealed through serum CEA monitoring that only a small fraction of tumor cells were infected, with negligible indications of significant viral replication or amplification in vivo.120 Reporter genes engineered to engage with radioactive tracers have been assessed in human investigations. Oncolytic herpes simplex viruses (HSVs) can be seen using PET using the HSV TK enzyme, which phosphorylates specific positron-emitting substrates, thus sequestering them within infected cells. This notion was exemplified in a clinical experiment utilizing HSV TK–based gene therapy for glioblastoma.121 Nonetheless, clinical validation for monitoring the spread of replication-competent oncolytic viruses remains outstanding. The gene that encodes the thyroidal sodium iodide symporter (NIS), which is important for concentrating radioactive iodide, has been integrated into the genomes of various oncolytic viruses, such as adenovirus, measles virus, vesicular stomatitis virus (VSV), HSV, and vaccinia virus. These engineered viruses have undergone assessment in preclinical research utilizing diverse radioisotopes (125I, 123I, 124I, and 99mTcO₄⁻) to track in vivo viral dissemination.
This adaptable NIS-based imaging methodology was recently corroborated in a clinical experiment, wherein 99mTcO₄-based SPECT/computed tomography (CT) imaging effectively monitored the intratumoral dissemination of an oncolytic adenovirus designed to express NIS.122 The strategy known as radiovirotherapy has shown that the therapeutic efficacy of an NIS-expressing oncolytic virus can be augmented by the administration of ¹³¹I, a radioisotope that emits high-energy beta particles, thereby intensifying localized radiation in the infected tumor tissue.123
Potential for local intravitreal/intraocular administration.
In ocular oncology, oncolytic virotherapy (OV) serves not only as a complement to ICI but also as a transformative approach that can alter the tumor-immune environment. The eye, historically regarded as an immune-privileged organ, inherently restricts T-cell activation and promotes immunological tolerance elements that account for the low efficacy of PD-1/PD-L1 and CTLA-4 inhibition in the treatment of uveal melanoma. OVs confront this immune-suppressive environment by transforming the ocular milieu from a state of immunological quiescence to one of active immune activity. OVs function as in situ vaccines by lysing tumor cells and releasing neoantigens, thereby enhancing epitope presentation and initiating systemic antitumor immunity that can target far metastatic locations, especially in the liver.124,125 Recent findings show that immunotherapeutic blockade of CD47 can potentiate virus-mediated immune activation and enhance neutralizing antibody responses, offering a dual advantage in oncolytic virotherapy.126 This strategy is innovative because of the synthetic engineering of OVs tailored for ocular malignancies. These viruses, containing immune-stimulatory transgenes such as IL-15 superagonists, bispecific T-cell engagers, or CRISPR-based gene-editing modules, can concurrently reduce tumor mass, reinstate HLA expression, and eradicate immunosuppressive checkpoints at the genetic level. These developments expand the role of oncolytic viruses beyond direct cytolysis, positioning them as multifunctional platforms capable of modulating local immune responses.127 The application of local intravitreal or intraocular injection adds a novel dimension to oncolytic virotherapy. Unlike systemic treatments, this targeted delivery method attains high tumor selectivity with minimum systemic damage, while facilitating immunological signal transmission to far metastatic locations. Potentially enabling localized intraocular tumors to contribute to broader systemic immune activation.128
OV is being investigated more and more as a possible part of multimodal immunotherapy approaches for eye cancers. It can be strategically integrated with ICIs to overcome inhibitory barriers, with adoptive T-cell treatment to amplify antigen-specific clones, and with nanomedicine-based delivery methods to accurately regulate spatial and temporal release kinetics. This comprehensive method enhances OV from a simple “checkpoint-supportive” function to a pivotal catalyst in a new paradigm of ocular immuno-oncology, wherein localized therapy triggers systemic immune orchestration and enduring worldwide tumor control.129
Bispecific antibodies and immune engagers
Antibody-based bispecific therapies have shown significant effectiveness in the treatment of hematologic malignancies, and promising advancements are currently being made in the development of BiTE-based approaches for solid tumors. Nonetheless, these therapies are intrinsically constrained by their reliance on surface-expressed antigens as targets. Conversely, over 90% of the human proteome is located intracellularly, rendering it unattainable by traditional antibody-based methods and highlighting the necessity for alternate ways to address intracellular targets.130,131 The appearance of intracellular antigens on major histocompatibility complex (MHC) molecules enables their recognition. T-cell receptors (TCRs), which inherently identify MHC–peptide complexes that reflect the intracellular antigenic landscape, are thus optimal candidates for targeting concealed epitopes. MHC class I molecules are generally present on almost all nucleated cells; however, their expression may be diminished or obscured in cancer cells and cells infected by specific viruses, enabling these cells to avoid immune surveillance.132 As a result, genetic and protein engineers have endeavored to create TCRs that replicate the essential structural and functional characteristics of antibodies, especially their elevated binding affinity, generally around three orders of magnitude superior to that of natural TCRs and their soluble nature.
Initial attempts to produce soluble TCRs consisting exclusively of the variable domains from both chains (Vα and Vβ) were ineffective, since these constructs demonstrated inadequate solubility and structural instability. This was mostly attributable to the exposure of hydrophobic residues, typically concealed within the cell membrane, resulting in misfolding and aggregation.133 Nonetheless, researchers have encountered numerous technical obstacles intrinsic to the structure and biophysics of the TCR, most notably, the difficulty of maintaining TCR chains in a soluble state while concurrently augmenting binding avidity.
Following years of refining, these endeavors resulted in the creation of the inaugural FDA-approved bispecific TCR-based treatment, tebentafusp. This chemical specifically targets the HLA-A*02:01-restricted tumor-associated antigen (TAA) gp100 in uveal melanoma, a common malignant ocular tumor, representing a significant advancement in the clinical application of soluble TCR therapies.134 This therapy is classified as an immune-mobilizing monoclonal T-cell receptor against cancer (ImmTAC). It is presently being assessed in a phase II/III clinical trial for nonocular melanoma (NCT05549297).
The ImmTAC platform combines an affinity-enhanced TCR with a humanized scFv targeting CD3, facilitating effective T-cell redirection to tumor cells. Despite their remarkable tumor-killing potency, ImmTACs exhibit a brief plasma half-life of only a few hours, requiring regular dosage to sustain therapeutic effectiveness. In addition to tebentafusp, several TCR-based T-cell engagers (TCEs) are now being developed, aimed against intracellular tumor-associated antigens, including MAGE-A4/8, MAGE-A1, and WT1,135 or surviving.136 A novel TCE has been developed, employing the ectodomains of the Vγ9δ2 TCR for tumor recognition, referred to as the gamma delta TCR anti-CD3 bispecific molecule (GAB). The Vγ9δ2 TCR identifies butyrophilin (BTN) family members, facilitating tumor detection that is not contingent upon mutational load, MHC restriction, or TAA expression. The primary drawback of the GAB platform currently resides in the diminished expression levels of these modified molecules, which constrain their therapeutic efficacy and scalability in clinical applications.137 This novel family of T-cell engagers offers a substantial possibility to target hitherto unattainable intracellular antigens that classic bispecific antibodies or CAR-T treatments cannot detect. In contrast, initiatives to target oncopeptide–MHC complexes utilizing the CAR-T platform have surfaced, leading to the notion of peptide-centric CARs, which broaden the applicability of CAR technology beyond surface antigens to the MHC-presented intracellular peptide repertoire.138 Nonetheless, the MHC complex remains the inherent and most suitable ligand for TCRs, maintaining its complete signaling capacities and excellent functional responsiveness. The principal hurdles that persist include preserving the transmembrane structure of TCRs and mitigating their comparatively low binding affinity in physiological circumstances.94,139
Recent advancements in genetic and protein engineering have created new opportunities for improving natural biomolecular interactions, and TCEs have also profited from these technological improvements, facilitating the development of more effective and stable therapeutic constructions (Table 2).140,141 Even in cases when checkpoint inhibitors are ineffective, bentafusp has the most consistent clinical effectiveness among UM trials. This discrepancy shows that, whereas UM has antigens, it lacks effective T-cell engagement pathways.142 TCR-bispecifics directly reroute T lymphocytes with high affinity while avoiding poor MHC expression.143 However, there are still drawbacks: deep hepatic metastases often react insufficiently, cytokine toxicity is still significant, and advantages are mostly limited to HLA-A*02:01 individuals. Furthermore, inconsistent durability findings across cohorts imply that microenvironmental resistance is yet unresolved.10 All of the research points to tebentafusp as a foundational treatment that must be used in conjunction with microenvironment-modifying agents, such as OVs or specific metabolic inhibitors, to transform partial responses into long-lasting remission.6 Collectively, these platforms should not be viewed as immediate replacements for checkpoint inhibition, but as translational tools designed to interrogate and overcome specific biological bottlenecks in uveal melanoma, thereby informing rational combination strategies rather than single-modality escalation.
Table 2.
Next-generation immunotherapies for uveal melanoma are transitioning from traditional vaccine methodologies to synthetic immune platforms, emphasizing mechanistic mapping, clinical outcomes, and future innovation potential
|
Modality
|
Core mechanism
|
Clinical signal
|
Challenges
|
Future directions
|
References
|
| Whole-cell vaccines |
UM cells engineered with MHC-II + CD80 prime CD4⁺ T cells |
Anecdotal responses; polyclonal CD4⁺ activity |
Limited data; allele match; weak persistence |
Neoantigen vaccines; combos with ICIs |
144
|
| DC vaccines |
mRNA-transfected DCs present UM antigens |
Phase II: DFS 34.5 mo; OS ~79% |
No proven superiority; trials ongoing |
Phase III RNA-DCs; optimized maturation |
145,146
|
| ACT (TIL/TCR-T) |
Expanded TILs or engineered TCR-T (PRAME, SLC45A2, MAGE-C2) |
Case regressions; early PRAME-TCR trials |
Complex, HLA-restricted, tumor escape |
Universal TCRs; multiplex targeting |
147
|
| CAR-T cells |
scFv-based receptors target UM antigens, HLA-independent |
HER2 CAR-T active preclinically; GD2 trial |
Infiltration, toxicity, short persistence |
Armored CAR-Ts; cytokine support |
148
|
| Oncolytic viruses |
Engineered OVs lyse tumors, expose neoantigens |
T-VEC, JX-594 active in melanoma/HCC; UM concept |
Dose limits; immune clearance; off-target risk |
IL-15/CRISPR-armed OVs; OV + ICI/ACT |
149
|
| TCR-bispecifics |
Soluble TCRs fused to CD3 scFv redirect T cells |
Tebentafusp FDA-approved; GAB bypasses MHC |
HLA restriction, short half-life; low BTN |
Next-gen TCEs (WT1/MAGE); γδ-TCR engagers; Fc-fusion |
150
|
Overcoming the challenges of ocular immune privilege
The concept of immune privilege originated from early observations that allografts could survive for extended periods when transplanted into certain anatomical sites, including the brain and the anterior chamber of the eye.151 The primary function of immune privilege is to protect fragile and highly sensitive cells within specific tissue microenvironments. This privilege can be compromised under specific pathological conditions such as infection, advanced diabetic retinopathy (DR), or age-related macular degeneration (AMD) permitting microorganisms in the local environment to circumvent additional systemic innate and adaptive immune responses. Alternatively,152 immune privilege may be undermined by the deterioration or degeneration of retinal pigment epithelium (RPE) cells, which function as the physical barrier sustaining this condition. Conversely, the immunological privilege that safeguards the eye from inflammatory harm may facilitate the onset of antiretinal autoimmunity, wherein peripheral tolerance mechanisms inadequately respond to ocular antigens.153 Recent single-cell and spatial transcriptomic studies suggest that ocular immune privilege operates as a dynamically regulated system rather than a purely static barrier. Multi-omic analyses indicate that immune tolerance and inflammatory signaling can coexist within the ocular environment through coordinated interactions between adaptive and innate immune components. As summarized in Fig. 2, the integrated immunogenomic landscape illustrates how cellular heterogeneity, clonotype expansion, and HLA-associated regulatory mechanisms contribute to the balance between immune quiescence and activation.
Fig. 2.
Cellular topology and clonotype architecture defining immune regulation within the ocular immune-privileged niche. This figure presents a systems-level yet structurally simplified map of immune organization in the ocular microenvironment, illustrating how immune privilege emerges from coordinated cellular compartmentalization, controlled clonal expansion, and transcriptionally constrained immune states rather than global immune suppression. (A) Single-cell embedding of ocular immune populations reveals discrete clustering of adaptive (CD4⁺ and CD8⁺ T cells) and innate (macrophages and dendritic cells) compartments, indicating preserved immune architecture under immune-privileged conditions. (B) Quantitative clonotype-size profiling across CD4⁺ and CD8⁺ subsets demonstrates selective, size-restricted clonal expansion, consistent with localized antigen sensing rather than widespread polyclonal activation. (C) Network-based representation of immune states identifies non-random transcriptional neighborhoods, highlighting structured crosstalk between adaptive and innate immune compartments instead of diffuse immune infiltration. (D) Clonotype-resolved single-cell analysis distinguishes CD4⁺- and CD8⁺-specific expansion dynamics, reflecting differential immune engagement under ocular constraints. (E) Density distributions of activation- and exhaustion-associated gene signatures indicate compartment-specific immune tuning, with restrained effector amplification. (F) Antigen-associated clonotypes show selective enrichment, demonstrating that ocular immune privilege limits but does not abolish antigen-driven T-cell responses. (G) Differential transcriptional profiling of ocular CD4⁺ T cells reveals concurrent regulatory, differentiation, and controlled activation programs. (H) Functional pathway enrichment analysis highlights preferential engagement of antigen processing, interferon signaling, and immune-regulatory pathways, while excluding broad inflammatory cascades. Collectively, this figure emphasizes organizational coherence over panel density, demonstrating that ocular immune privilege is maintained through structured cellular topology and clonotype governance that permit immune surveillance while preventing uncontrolled effector escalation. Panels were intentionally curated and hierarchically organized to enhance interpretability and mechanistic clarity.154 Reprinted from Concepcion. et al (distributed under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
.
Cellular topology and clonotype architecture defining immune regulation within the ocular immune-privileged niche. This figure presents a systems-level yet structurally simplified map of immune organization in the ocular microenvironment, illustrating how immune privilege emerges from coordinated cellular compartmentalization, controlled clonal expansion, and transcriptionally constrained immune states rather than global immune suppression. (A) Single-cell embedding of ocular immune populations reveals discrete clustering of adaptive (CD4⁺ and CD8⁺ T cells) and innate (macrophages and dendritic cells) compartments, indicating preserved immune architecture under immune-privileged conditions. (B) Quantitative clonotype-size profiling across CD4⁺ and CD8⁺ subsets demonstrates selective, size-restricted clonal expansion, consistent with localized antigen sensing rather than widespread polyclonal activation. (C) Network-based representation of immune states identifies non-random transcriptional neighborhoods, highlighting structured crosstalk between adaptive and innate immune compartments instead of diffuse immune infiltration. (D) Clonotype-resolved single-cell analysis distinguishes CD4⁺- and CD8⁺-specific expansion dynamics, reflecting differential immune engagement under ocular constraints. (E) Density distributions of activation- and exhaustion-associated gene signatures indicate compartment-specific immune tuning, with restrained effector amplification. (F) Antigen-associated clonotypes show selective enrichment, demonstrating that ocular immune privilege limits but does not abolish antigen-driven T-cell responses. (G) Differential transcriptional profiling of ocular CD4⁺ T cells reveals concurrent regulatory, differentiation, and controlled activation programs. (H) Functional pathway enrichment analysis highlights preferential engagement of antigen processing, interferon signaling, and immune-regulatory pathways, while excluding broad inflammatory cascades. Collectively, this figure emphasizes organizational coherence over panel density, demonstrating that ocular immune privilege is maintained through structured cellular topology and clonotype governance that permit immune surveillance while preventing uncontrolled effector escalation. Panels were intentionally curated and hierarchically organized to enhance interpretability and mechanistic clarity.154 Reprinted from Concepcion. et al (distributed under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
Classical mechanisms of ocular immune privilege
The retina, functioning as an extension of the brain, is an immune-privileged region within the eye.155-157 The immune privilege mechanism evolved to safeguard retinal integrity and avert inflammatory damage, protecting the retina's very sensitive neurons that have restricted regenerative and reparative capabilities. Multiple publications have offered extensive reviews on ocular immune privilege, including its historical context, fundamental mechanisms, and practical significance, especially for corneal transplantation.158-162 The concept of an immunologically privileged region in the eye, specifically the anterior chamber, was initially presented by Medawar in 1948.163 Numerous immunological cells, biochemical pathways, and regulatory factors are recognized for their roles in sustaining ocular immune privilege. This safeguarding is accomplished by both active and passive mechanisms:
BRB and blood–aqueous barrier function not only as anatomical boundaries but also as active regulators of immune access, restricting leukocyte trafficking and limiting the intraocular penetration of systemically delivered immunotherapeutic agents. 1) Diverse endothelial cells, immunological cells (such as natural killer and dendritic cells), and retinal neurons establish a secondary defense by releasing immunosuppressive substances that inhibit effector T-cell activity and restrict intraocular inflammation. 2) Antigen entry into the anterior chamber, vitreous cavity, or subretinal space induces ACAID, a tolerogenic process characterized by systemic suppression of delayed-type hypersensitivity and preferential expansion of regulatory T-cell populations, thereby limiting durable effector T-cell responses against intraocular tumors. This phenomenon is marked by significant immune suppression facilitated by CD4⁻CD8⁺ regulatory T cells and the modulation of Th1- and Th2-mediated inflammatory responses, thereby forming a tertiary layer of immune defense.153,164 This summary emphasizes the essential physiological and molecular mechanisms that support ocular immune privilege. The BRB is crucial in restricting the entry of circulating immune cells into the eye. The structure comprises two elements: the outer BRB, constituted by the tight connections of RPE cells, and the inner BRB, produced by retinal capillary endothelial cells.165,166 The outer BRB facilitates immune privilege by secreting substances such as pigment epithelial-cell-derived factor and vascular endothelial growth factor (VEGF), which maintain the structural and functional integrity of the retinal vasculature and choriocapillaris.167,168 The inner blood-retinal barrier consists of pericytes, astrocytes, Müller cells, the collagen IV basement membrane, and perivascular macrophages.169,170 The anterior ocular barrier, referred to as the blood–aqueous barrier, comprises vascular endothelial cells in the iris and nonpigmented ciliary epithelial cells, serving to inhibit the movement of chemicals from plasma to the aqueous humor.171,172 The disruption of these physical barriers undermines immunological privilege and may result in retinal disease, marked by increased BRB permeability and heightened capillary hydrostatic pressure in the eye.173,174 When physical barriers are breached, innate and adaptive immune responses in the intraocular microenvironment are activated, leading to immune modulation and inflammatory suppression mediated by effector immune cells and endothelial cells. The immunoregulatory action is mediated by soluble components in the aqueous humor and membrane-bound compounds from other intraocular compartments. Principal soluble mediators encompass transforming growth factor beta (TGF-β), alpha-melanocyte-stimulating hormone, calcitonin gene-related peptides, thrombospondin-1, vasoactive intestinal peptides, and complement-inhibitory compounds.175 Aqueous humor is a translucent, colorless fluid found in the anterior and posterior chambers of the eye.176 The aqueous humor, an essential element of the eye, contains many immunomodulatory substances such as neuropeptides, cytokines, growth factors, and soluble cell-surface receptors, which together inhibit inflammatory responses and promote the activation of regulatory T cells.158,177,178 Consequently, the innate and adaptive immune responses operate to mitigate inflammation, augment the generation of anti-inflammatory cytokines, and facilitate the activity of immunological-tolerance effector T cells and other immune effector cells.179-183 Intraocular resident immune cells, including macrophages and microglia, participate in active neuroimmune modulation through interactions with retinal neurons and glial cells, producing neuropeptides and inhibitory cytokines that suppress local inflammation. While essential for retinal protection, this neuroimmune regulation further constrains antitumor immune activation and contributes to resistance against immune checkpoint–based therapies. Antigens located in the anterior chamber can elicit systemic immune suppression targeting specific infections.184 This effect is also observed in the vitreous cavity and subretinal space.185,186 Retinal resident microglia behave as antigen-presenting cells with diminished MHC class II expression, functioning as an immune checkpoint by suppressing T-cell activation and fostering anti-inflammatory responses.175,187 Ocular immune privilege exerts both protective and permissive effects within the ocular microenvironment. It establishes a microenvironment that is separated from systemic circulation, protecting all elements of the visual system; even little disturbances can result in significant ocular illnesses and jeopardize the effectiveness of ocular allografts.162 Conversely, the rigidity of immune privilege may constitute a weakness, as demonstrated by the immunological tolerance of intraocular malignancies and the onset of infectious blindness resulting from illnesses such as trachoma, river blindness, or herpes simplex virus infection.159,188 Addressing ocular immune privilege requires more than simply shifting between immune suppression and activation; it depends on carefully controlled temporal and spatial modulation; it requires temporal and spatial reconfiguration. Next-generation ocular immunotherapies aim to achieve reversible and localized immune modulation, enabling therapeutic intervention while preserving the protective features of immune privilege. As shown in Fig. 3, ocular immune privilege is reinforced by HLA-structured transcriptional immune states that permit surveillance while restricting effector activation.
Fig. 3.
HLA-structured immune-state organization sustaining ocular immune privilege. This figure provides a condensed, mechanism-oriented representation of how ocular immune privilege is maintained through coordinated cellular, clonal, and transcriptional programs rather than diffuse immune suppression. (A) Relative distribution of resting and effector CD4⁺ and CD8⁺ T-cell subsets highlights restrained effector representation under immune-privileged conditions. (B) Clone-size stratification reveals selective, non-random clonal expansion patterns, indicating controlled antigen engagement rather than broad polyclonal activation. (C) Expression mapping of activation, differentiation, and effector-lineage markers (including TH1/TH17-associated programs) demonstrates tightly regulated functional polarization. (D–F) Single-cell clonotype analyses distinguish CD4⁺ and CD8⁺ T-cell expansion dynamics and identify antigen-associated clonotypes without widespread inflammatory amplification. (G) Differential gene-expression profiling of ocular CD4⁺ T cells highlights an HLA-associated transcriptional axis integrating immune regulation, interferon responsiveness, and limited effector activation. (H) Pathway-level enrichment analysis confirms preferential engagement of antigen processing, interferon signaling, and immune-regulatory pathways while excluding broad inflammatory cascades.154 Collectively, the figure emphasizes organizational clarity over panel density, illustrating that ocular immune privilege is reinforced by HLA-structured immune states that permit immune surveillance while constraining effector escalation. Panels were intentionally curated and visually simplified to enhance interpretability and reduce redundancy, enabling direct mechanistic insight into immune-privileged regulation. Reprinted from Concepcion. et al (distributed under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
.
HLA-structured immune-state organization sustaining ocular immune privilege. This figure provides a condensed, mechanism-oriented representation of how ocular immune privilege is maintained through coordinated cellular, clonal, and transcriptional programs rather than diffuse immune suppression. (A) Relative distribution of resting and effector CD4⁺ and CD8⁺ T-cell subsets highlights restrained effector representation under immune-privileged conditions. (B) Clone-size stratification reveals selective, non-random clonal expansion patterns, indicating controlled antigen engagement rather than broad polyclonal activation. (C) Expression mapping of activation, differentiation, and effector-lineage markers (including TH1/TH17-associated programs) demonstrates tightly regulated functional polarization. (D–F) Single-cell clonotype analyses distinguish CD4⁺ and CD8⁺ T-cell expansion dynamics and identify antigen-associated clonotypes without widespread inflammatory amplification. (G) Differential gene-expression profiling of ocular CD4⁺ T cells highlights an HLA-associated transcriptional axis integrating immune regulation, interferon responsiveness, and limited effector activation. (H) Pathway-level enrichment analysis confirms preferential engagement of antigen processing, interferon signaling, and immune-regulatory pathways while excluding broad inflammatory cascades.154 Collectively, the figure emphasizes organizational clarity over panel density, illustrating that ocular immune privilege is reinforced by HLA-structured immune states that permit immune surveillance while constraining effector escalation. Panels were intentionally curated and visually simplified to enhance interpretability and reduce redundancy, enabling direct mechanistic insight into immune-privileged regulation. Reprinted from Concepcion. et al (distributed under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
Re-engineering immune privilege: localized and reversible modulation
How immunotherapies are administered, as opposed to which immunological targets are chosen, is a crucial but sometimes disregarded factor in precision immune engineering in ocular cancer.189 The therapeutic environment in the eye is particularly limited because intraocular volume is constricted, molecular trafficking is restricted by the blood–retinal and blood–aqueous barriers, and even little inflammatory damage may permanently impair vision.190 In this situation, systemic immune agent delivery often turns out to be ineffective or clinically undesirable. Biodegradable nanoparticles, responsive nanocarriers,190 and injectable hydrogel depots are examples of nanotechnology-based delivery systems that provide a way to balance ocular safety with immune activation.191 In response to microenvironmental signals like hypoxia, acidity, or inflammatory signaling, these systems may influence immune function in a geographically and temporally limited way by facilitating local drug retention and stimulus-dependent release.192 For drugs with limited therapeutic windows, localized delivery methods, specifically, intravitreal and intraocular administration, have grown in significance as a means of implementing precision immunological tactics in ocular treatment.193 According to this viewpoint, delivery engineering is a crucial component of the design of precision immunotherapy for the eye rather than only being supportive.193-195 This distinction is critical, as it separates immune-engineering approaches designed for ocular tumors from systemic immunotherapy paradigms that do not account for spatial and anatomical constraints.189 One method employs nanoparticle-encoded immune modulators that specifically deliver short-acting inhibitors of TGF-β2, α-MSH, or IDO in hypoxic tumor environments. Such nanoparticle-based systems are designed to create localized regions of transient immune modulation, allowing effector cell access while maintaining overall ocular immune tolerance. By designing particles that respond to reactive oxygen species or matrix metalloproteinases, immunosuppression is alleviated solely in areas determined by tumor physiology.196 Adaptive biomaterials represent an emerging approach in ocular immunotherapy. Intelligent hydrogels infused with checkpoint inhibitors or cytokine agonists can modulate their release in response to intraocular signals such as pH variations, lactate buildup, or angiogenic factors, guaranteeing that immune regulation is localized to tumor-afflicted areas. Certain designs integrate self-healing polymers, allowing the hydrogel to reseal and reestablish immunological tolerance when the therapeutic payload is exhausted. Energy-based approaches have also been explored as non-pharmacological methods to transiently enhance ocular drug and immune-cell delivery.197 Pulsed focused ultrasound techniques, in conjunction with microbubbles, can temporarily disrupt the blood-retina barrier for many hours, allowing the penetration of bispecific antibodies, CAR-T cells, or NK cell engagers. Likewise, photothermal nanorods optimized for near-infrared wavelengths can accurately “unlock” tight connections with micron-scale precision.197 Synthetic immune engineering is at the forefront. Logic-gated bispecific antibodies that require concurrent identification of a tumor antigen (e.g., gp100) and a stress signal (e.g., hypoxia-induced CAIX) guarantee that immune activation is restricted to malignant locations. Simultaneously, CRISPR-based temporal editing facilitates the temporary suppression of immunosuppressive ligands like FasL or PD-L1 on retinal pigment epithelium, succeeded by their regulated re-expression post-therapy to restore tissue homeostasis.198 This perspective reframes immune privilege as a dynamically regulated biological system that can be transiently modified and subsequently restored to preserve ocular homeostasis. Such immuno-engineering strategies may enable effective tumor control while minimizing disruption to the eye’s inherent immunological protective mechanisms (Fig. 4).199
Fig. 4.
Simplified visualization of HLA-associated immune-state organization in the ocular microenvironment.This updated illustration shows a smaller, more concentrated picture of the eye's immune system to make it easier to understand and read. The study combines all the important parts into one clear picture that shows how the immune system is organized in space and what it means for its function, without using separate panels. UMAP embedding of ocular immune cells, color-coded by correlation with HLA-B27–associated uveitis, illustrates that cells with analogous association scores aggregate into distinctly separated transcriptional neighborhoods, signifying a non-random spatial organization of immune states rather than diffuse or overlapping patterns. Functional pathway enrichment of immune states linked to HLA-B27 shows that antigen processing and presentation, interferon signaling, the innate immune response, and immune-regulatory processes are all very important. This shows that immune activation is pathway-specific and controlled in the immune-privileged ocular environment. This picture emphasizes readability and conceptual coherence by reducing superfluous visual segmentation and boosting visual continuity. This makes it easier to understand HLA-linked immune organization.154 Reprinted from Concepcion. et al(distributed under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
.
Simplified visualization of HLA-associated immune-state organization in the ocular microenvironment.This updated illustration shows a smaller, more concentrated picture of the eye's immune system to make it easier to understand and read. The study combines all the important parts into one clear picture that shows how the immune system is organized in space and what it means for its function, without using separate panels. UMAP embedding of ocular immune cells, color-coded by correlation with HLA-B27–associated uveitis, illustrates that cells with analogous association scores aggregate into distinctly separated transcriptional neighborhoods, signifying a non-random spatial organization of immune states rather than diffuse or overlapping patterns. Functional pathway enrichment of immune states linked to HLA-B27 shows that antigen processing and presentation, interferon signaling, the innate immune response, and immune-regulatory processes are all very important. This shows that immune activation is pathway-specific and controlled in the immune-privileged ocular environment. This picture emphasizes readability and conceptual coherence by reducing superfluous visual segmentation and boosting visual continuity. This makes it easier to understand HLA-linked immune organization.154 Reprinted from Concepcion. et al(distributed under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
Drug delivery systems and localized administration in ocular immunotherapy
Systemic delivery of immunotherapeutic agents for ocular malignancies is often limited by the BRB and blood–aqueous barrier (BAB), which restricts intraocular drug penetration and may increase the risk of systemic immune-related adverse effects. Intravitreal injection remains the clinical standard for direct ocular drug administration, allowing bypass of the blood–retinal barrier and achievement of high local drug concentrations.199 However, repeated intravitreal injections carry cumulative risks, including infection and retinal detachment. Next-generation strategies, such as protease-activated “split” antibody constructs, are designed to enable preferential activation within the tumor microenvironment, thereby reducing off-target effects.199 Nanoparticle-based carriers are increasingly being explored for localized ocular immunotherapy. Platforms including liposomes, dendrimers, polymeric micelles, and exosome-like vesicles have been engineered to enhance trans corneal and transretinal delivery, enable sustained drug release, and limit systemic exposure.200 Nanocarriers designed for uveal melanoma and retinoblastoma exhibit the capability to attain elevated intraocular bioavailability while minimizing systemic medication exposure.201 Biodegradable nanoparticles further support this approach by offering controlled release profiles adapted to local ocular physiology.190 Hydrogels offer a means of localized drug delivery with improved spatial and temporal control. Injectable hydrogels capable of co-encapsulating drug-loaded nanoparticles can form in situ depots within the vitreous, enabling prolonged and localized drug release.202 Responsive hydrogel networks that react to local cues such as acidity, reactive oxygen species (ROS), or hypoxia, initially developed for cancer immunotherapy, provide a useful design framework for ocular applications.203 Immunostimulatory peptide-based hydrogels may further support localized drug delivery while modulating the ocular immune environment and limiting systemic immune-related adverse effects (Table 3).204 This reframing of ocular immune privilege as a dynamically tunable system rather than a fixed barrier provides the conceptual bridge between mechanistic understanding and biomarker-guided precision immunotherapy discussed in the following section.
Table 3.
Reprogramming ocular immune privilege: novel techniques transforming obstacles into therapeutic conduits
|
Strategy
|
Mechanistic concept
|
Current limitations
|
Innovative advances
|
References
|
| Barrier modulation |
BRB and BAB act as physical gates to immune infiltration |
Breach risks vision loss; systemic delivery is blocked |
Ultrasound + microbubbles; NIR nanorods for transient, localized barrier loosening |
205
|
| Microenvironment reprogramming |
Aqueous humor and RPE secrete TGF-β, α-MSH, IDO, suppressing effector T-cells |
Global immunosuppression hinders anti-tumor responses |
Nanoparticles releasing short-acting inhibitors only in hypoxic tumor niches |
206
|
| Smart biomaterials |
Hydrogels or adaptive polymers release drugs in response to intraocular cues. |
Current systems lack dynamic control, risk of chronic inflammation |
Self-healing hydrogels tuned to pH/ROS/lactate; checkpoint-loaded adaptive biomaterials |
207
|
| Synthetic immune engineering |
Logic-gated antibodies, CRISPR-based ligand silencing for tumor-specific activation |
Lack of reversible control; off-target systemic activation |
Dual-input ImmTACs (antigen + stress signal); temporal CRISPR switches for FasL/PD-L1 |
208
|
| Localized delivery systems |
Intravitreal injections, nanoparticles, and hydrogels bypass systemic circulation. |
Frequent injections carry infection/retinal detachment risk |
Protease-activated antibodies; biodegradable nanocarriers; 3D hydrogels as long-term depots |
209,210
|
Biomarkers and precision immunotherapy
Recent proteomic studies have contributed to improved prognostic assessment in patients with UM. A recent large-scale study using LC–MS/MS iTRAQ-based proteomics analyzed primary UM samples from 53 metastasizing and 47 non-metastasizing patients.211 A total of about 3,935 proteins were evaluated across all samples, with bioinformatics analysis revealing 191 proteins increased in metastatic patients and 211 proteins upregulated in non-metastatic instances.211 Reactome pathway analysis of proteins enriched in metastatic UM revealed overrepresentation of immune-related pathways, as well as pathways associated with vesicle-mediated transport, extracellular matrix organization, and protein metabolism.211 In contrast, non-metastatic patients had a predominance of pathways related to metabolism, cellular responses to environmental stimuli, and developmental biology.211 Immune system-related proteins were significantly overrepresented in metastatic patients, while housekeeping pathways exhibited stronger enrichment in non-metastatic instances.211 These findings support the presence of an immunosuppressive phenotype in primary UM, characterized by relatively low expression of immune checkpoint–related molecules.211 Nevertheless, certain molecules such as CDH1 and HLA-DPA1, in addition to 15 kinases and phosphatases, have been identified as promising novel targets for immune checkpoint suppression therapy.211 Using these data, the authors proposed a predictive model based on 32 proteins, which achieved an accuracy of approximately 93% in identifying patients at risk of metastatic progression.211 Such studies may inform the future development of immunoassays for non-invasive UM detection using blood or other biological samples.212-214 Subsequent research with bigger cohorts of UM patients will be crucial for a more precise evaluation of the predictive capabilities of protein profiles obtained from primary UM proteomics. In parallel, liquid biopsy approaches based on circulating tumor DNA (ctDNA) are increasingly being used for non-invasive cancer monitoring. A study involving 69 MUM patients treated with tebentafusp revealed that detectable ctDNA at baseline indicated a worse prognosis, while a ≥90% reduction in ctDNA after 12 weeks was linked to significantly enhanced survival, nearing the outcomes of patients with initially undetectable ctDNA.215 These findings indicate that ctDNA may serve as an early and sensitive biomarker for monitoring treatment response and guiding therapeutic decisions.
Aqueous humor (AH) liquid biopsy represents a minimally invasive approach for molecular analysis in retinoblastoma (RB), a disease in which conventional tumor biopsies are generally avoided because of procedural risks. Tumor-derived cell-free DNA can be obtained from aqueous humor samples collected via ocular paracentesis, enabling genomic and epigenetic analyses.216 The DNA methylation profiles in AH-derived cfDNA closely resemble those of primary RB tumors, encompassing significant modifications such as RB1 promoter hypermethylation and MYCN/SYK promoter hypomethylation, thereby facilitating precise molecular categorization without enucleation.217 Serial aqueous humor sampling allows longitudinal assessment of tumor molecular features and has been associated with treatment outcome prediction in retinoblastoma.216 Imaging biomarkers, including optical coherence tomography (OCT) and positron emission tomography (PET), have expanded the ability to monitor ocular tumors and immune-related changes in vivo.218-220 OCT, providing micrometer-scale resolution, has historically been utilized for the structural imaging of retinal and choroidal malignancies; nevertheless, its nascent function as a proxy for immunological activation is just recently gaining recognition.221,222 OCT angiography (OCT-A) can identify immunotherapy-induced vascular remodeling, localized edema due to immune infiltration, and microvascular pruning in regressing malignancies. Alterations in artery density and flow signals may function as non-invasive markers of T-cell activity and cytokine-mediated inflammation. These structural changes may indirectly reflect immune-related activity within ocular tissues.223 Conversely, PET offers a molecular insight into immunological processes. Innovative tracers, like granzyme B–specific probes, can assess cytotoxic T-cell degranulation, whereas galectin-1 ligands assist in forecasting responses to checkpoint blockage. Immuno-PET approaches exceed conventional FDG-PET by differentiating genuine tumor development from pseudoprogression, a vital distinction in ocular malignancies, where edema or fluid accumulation may otherwise resemble disease exacerbation.224 When integrated with radiomics, PET data produce intricate textural characteristics linked to CD8⁺ T-cell infiltration and immunological heterogeneity, offering predictive insights that surpass the capabilities of histology alone. The combined use of OCT and PET enables complementary assessment of localized ocular changes and systemic immune activity: OCT acts as a localized, high-resolution indication of structural and vascular immune alterations in the eye, whereas PET serves as a systemic, molecular monitor of effector immune activity.225 Together, these modalities allow correlation of local ocular tissue changes with systemic immune markers, supporting more informed evaluation of therapeutic response. This method enables prompt evaluation of therapy effectiveness, guides eye-preserving strategies, and aids in preventing excessive treatment. Imaging-based immune monitoring may contribute to more precise and individualized management of ocular malignancies by enabling longitudinal, non-invasive assessment of treatment response (Table 4).146 Collectively, these findings reveal that uveal melanoma maintains an activeimmuneprivilege through a multilayered network of genetic rearrangements, cytokine signaling, metabolic reprogramming, and intercellular interactions. Rather than a state of passive tolerance, it represents a dynamicimmunosuppressive ecosystem that continually recalibrates its inhibitory and escape circuits. The schematic below Fig. 5 summarizes these interwoven processes from BAP1-driven transcriptional reprogramming and cytokine-mediated tolerance to the cross-communication among immune and stromal cell populations, establishing the biological foundation for next-generation therapeutic paradigms. These include combination strategies, microbiome-based interventions, and AI-enabled immunotherapies discussed in the subsequent sections.
Table 4.
Innovative biomarker approaches in ocular oncology: integrating molecular, liquid, and imaging techniques to enhance precision immunotherapy.
|
Biomarker modality
|
Core concept
|
Clinical/preclinical signal
|
Challenges
|
Future directions
|
References
|
| Proteomic Signatures |
Protein atlases stratify metastatic vs. non-metastatic UM |
The 32-protein panel predicted metastasis with 93% accuracy |
Small cohorts; clinical integration |
AI-based risk scores; blood proteo-immunoassays |
211
|
| ctDNA |
Plasma mutation tracking for real-time monitoring |
≥90% reduction after tebentafusp → improved survival |
Low sensitivity in small tumors |
Adaptive therapy; minimal residual disease detection |
226
|
| Aqueous Humor Biopsy |
cfDNA from AH as a molecular window into tumors |
RB1, MYCN, and SYK methylation predicted outcomes in RB |
Invasive; limited sample volume |
AH classifiers guiding eye salvage vs enucleation |
227
|
| OCT / OCT-A |
Imaging immune effects via vascular/tissue remodeling |
Shows pruning, edema, and infiltration linked to T-cell activity |
Mostly structural, not molecular |
OCT “immunograms” linking vasculature + immune cues |
228,229
|
| PET Imaging |
Tracers visualize systemic immune activity |
Granzyme B, galectin-1 PET predict immunotherapy response |
Radiation; limited ocular resolution |
Immuno-PET + radiomics immune fingerprints |
230
|
| OCT + PET |
Integrated local + systemic immune monitoring |
Validates ocular remodeling with systemic T-cell activity |
Cost: multimodal integration |
Dynamic immune atlases replacing static biopsy |
231
|
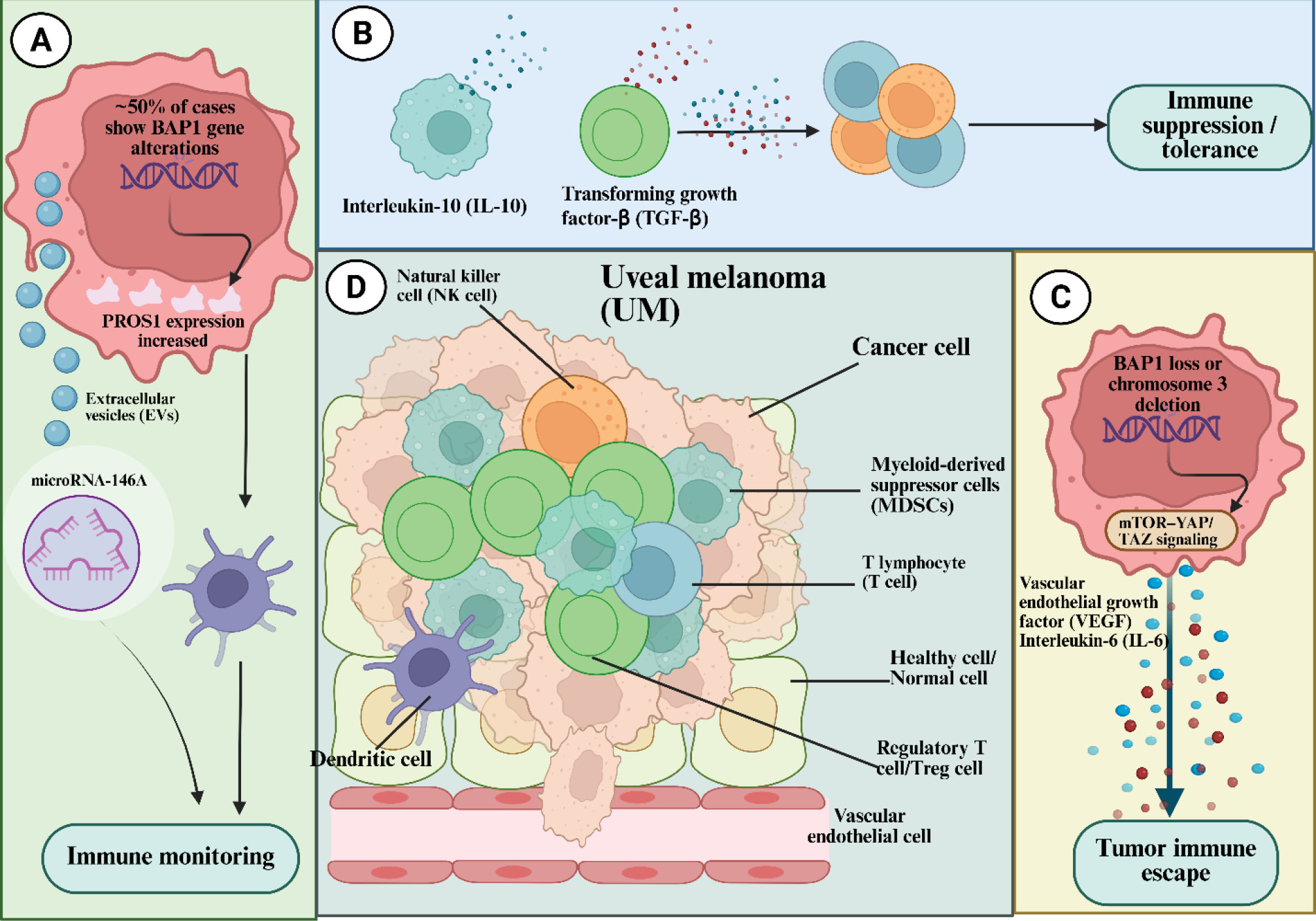
Fig. 5.
Multilayered immune-evasion network orchestrating uveal-melanoma persistence within the immune-privileged ocular niche. This composite schematic delineates the sequential transition from immune surveillance to tolerance and ultimately full immune escape in uveal melanoma, integrating genetic, cytokine, and cellular dimensions of tumor-immune crosstalk. (A) Genetic and vesicular priming phase: Approximately half of uveal-melanoma cases exhibit BAP1 loss, triggering aberrant PROS1 signaling and release of extracellular vesicles (EVs) enriched with microRNA-146A, which modulate dendritic-cell antigen presentation and suppress the initiation of cytotoxic responses. (B) Cytokine-mediated tolerance stage: Tumor and stromal compartments secrete high levels of IL-10 and TGF-β, reshaping the ocular milieu toward a tolerogenic phenotype that expands regulatory T cells (Tregs) and inhibits antigen-presenting functions. (C) Metabolic and angiogenic escape axis: Loss of chromosome 3/BAP1 activates the mTOR–YAP/TAZ signaling cascade, up-regulating VEGF and IL-6, which couple angiogenesis to immune evasion through endothelial-barrier modulation and MDSC recruitment. (D) Cellular ecosystem of sustained suppression: Within the uveal-melanoma microenvironment, cross-communication among NK cells, T lymphocytes, Tregs, MDSCs, dendritic cells, and endothelial cells establishes a feedback-locked immunosuppressive network that maintains ocular immune privilege and allows long-term tumor persistence despite systemic immunity.6 Adapted from Song et al. (distributed under the terms of the Creative Commons Attribution License (CC BY).
.
Multilayered immune-evasion network orchestrating uveal-melanoma persistence within the immune-privileged ocular niche. This composite schematic delineates the sequential transition from immune surveillance to tolerance and ultimately full immune escape in uveal melanoma, integrating genetic, cytokine, and cellular dimensions of tumor-immune crosstalk. (A) Genetic and vesicular priming phase: Approximately half of uveal-melanoma cases exhibit BAP1 loss, triggering aberrant PROS1 signaling and release of extracellular vesicles (EVs) enriched with microRNA-146A, which modulate dendritic-cell antigen presentation and suppress the initiation of cytotoxic responses. (B) Cytokine-mediated tolerance stage: Tumor and stromal compartments secrete high levels of IL-10 and TGF-β, reshaping the ocular milieu toward a tolerogenic phenotype that expands regulatory T cells (Tregs) and inhibits antigen-presenting functions. (C) Metabolic and angiogenic escape axis: Loss of chromosome 3/BAP1 activates the mTOR–YAP/TAZ signaling cascade, up-regulating VEGF and IL-6, which couple angiogenesis to immune evasion through endothelial-barrier modulation and MDSC recruitment. (D) Cellular ecosystem of sustained suppression: Within the uveal-melanoma microenvironment, cross-communication among NK cells, T lymphocytes, Tregs, MDSCs, dendritic cells, and endothelial cells establishes a feedback-locked immunosuppressive network that maintains ocular immune privilege and allows long-term tumor persistence despite systemic immunity.6 Adapted from Song et al. (distributed under the terms of the Creative Commons Attribution License (CC BY).
Building on this immunological framework, the following section transitions from mechanistic insights to translational innovation, outlining how integrative strategies combining radiotherapy, targeted therapy, epigenetic modulation, and intelligent immune engineering can reprogram ocular immune privilege and overcome the persistent barriers to effective tumor eradication.
Future horizons and translational directions
Combination therapies: checkpoint inhibitors with radiation, targeted therapy, or epigenetic modulators.
Despite advances in systemic immuno-oncology, ocular malignancies, particularly UM and CM, remain largely resistant to ICI monotherapy. This resistance has prompted investigation of combination strategies that integrate ICIs with radiotherapy, targeted therapies, or epigenetic modulators to enhance tumor immune responsiveness.232
Radiation + ICIs: amplifying the abscopal effect
Radiation therapy (RT) induces tumor cell death and can promote the release of tumor-associated antigens and immunogenic signals that support T-cell priming. When combined with immune checkpoint inhibitors, radiotherapy can elicit systemic anticancer responses through the abscopal effect. A 2025 retrospective analysis involving 24 metastatic uveal melanoma patients treated with stereotactic radiation combined with ICIs reported an overall response rate of 39.1%, a median PFS of 11.6 months, and an OS of 27.6 months, indicating a significant enhancement compared to prior results with ICI alone.233 These findings suggest that radio-immunotherapy may enhance immune activity in a subset of UM tumors compared with ICI monotherapy.
Targeted therapy + ICIs: exploiting MAPK pathway alterations
In contrast to UM, CM often contains mutations in the MAPK signaling pathway, particularly in BRAF, NRAS, and NF1, mirroring the mutation pattern observed in cutaneous melanoma. This molecular profile provides a rationale for evaluating combinations of BRAF/MEK inhibitors with ICIs, particularly in conjunctival melanoma. Preclinical models and initial clinical investigations indicate that targeted therapy can enhance tumor antigen presentation and inhibit immunosuppressive mechanisms, therefore augmenting tumor responsiveness to checkpoint inhibition.234 Although not yet standard practice, such combinations may offer clinical benefit in selected patient populations.
Epigenetic modulators + ICIs: a novel frontier
The combination of ICIs with epigenetic modulators, including HDAC inhibitors and DNA methyltransferase inhibitors, represents a highly unique treatment strategy. These drugs can reinstate antigen presentation by augmenting MHC expression, reactivating dormant tumor-associated antigens, and decreasing immunosuppressive myeloid cells. Although direct data on ocular malignancies is scarce, preclinical models and initial clinical trials in other solid tumors suggest that epigenetic remodeling may transform immunologically-desert UM lesions into forms sensitive to ICI. This approach focuses on modifying tumor–immune interactions through epigenetic regulation rather than solely intensifying immune activation.
Role of microbiome-immune interactions in ocular oncology
Recent studies suggest that microbiome–immune interactions may influence tumor development, immune evasion, and treatment response in ocular malignancies. Despite the eye's historical classification as an immune-privileged organ with limited microbial presence, recent metagenomic sequencing has uncovered distinct microbial populations on the ocular surface, including Corynebacterium, Staphylococcus, and Cutibacterium, which can modulate local immune responses and modify the immunologic microenvironment.235 These microbial communities engage with the ocular mucosal immune system through pattern recognition receptors (PRRs), particularly TLR2 and TLR4, which subsequently regulate downstream cytokine signaling pathways, change antigen presentation, and affect T-cell activation within the local immunological network.236 In ocular surface squamous neoplasia (OSSN) and conjunctival melanoma, microbiome-immune interactions can have multiple effects, either augmenting immune surveillance or facilitating immune evasion via chronic inflammatory signaling and Treg activation. Recent evidence suggests that commensal microorganisms can influence tumor immune checkpoints by modifying PD-L1 expression and MHC-I presentation on tumor and stromal cells, thereby diminishing cytotoxic T lymphocyte (CTL) infiltration and undermining the overall effectiveness of immunotherapy.237 Specific bacterial species produce short-chain fatty acids (SCFAs) and histone deacetylase inhibitors (HDACi) that can epigenetically modulate tumor suppressor genes or stimulate pro-tumorigenic cytokine promoters in ocular tissues. Furthermore, the gut–ocular microbiome axis, also known as the gut–retina link, has been demonstrated to affect systemic immune responses pertinent to intraocular malignancies, such as uveal melanoma.238 Experimental concepts such as ocular microbiome modulation, modeled after fecal microbiota transplantation, have been proposed but remain largely theoretical. Advances in synthetic biology have led to experimental exploration of engineered microbial systems for localized immunomodulation.239 Advanced 3D ocular tumor organoid systems, when co-cultured with microbiota, provide real-time observation of microbe–immune–tumor interactions, offering a robust experimental model for therapeutic evaluation, mechanistic investigation, and tailored immunotherapy response forecasting.239 The ocular microbiome has transitioned from a perceived passive entity to an active regulator of immunological equilibrium and tumor development. Understanding the interaction among the microbiome, immune system, and tumor may reveal novel diagnostic indicators, predictive biomarkers, and advanced microbiome-based immunotherapeutic approaches in ocular malignancy. Emerging evidence suggests that non-coding RNAs play critical regulatory roles in immune signaling, antigen presentation, and cellular differentiation, highlighting their potential as modulators of tumor immunogenicity and therapeutic response.240 Many of the underlying computational techniques are currently being actively used in modern cancer immunology, even if the ideas presented above reflect forward-looking and mostly theoretical applications of artificial intelligence within ocular immunotherapy.241 In this regard, the methodological underpinnings for the ultimate development of more adaptable and closed-loop AI-driven immune systems are provided by recent developments in computational immunology and machine learning.241 In order to bridge the gap between theoretical immune engineering notions and current translational practice, the next part focuses on existing and new computational methodologies that are currently affecting immunotherapy design, patient stratification, and treatment optimization.241
Artificial intelligence
Artificial intelligence (AI) has been increasingly applied in ocular oncology to support data integration, pattern recognition, and treatment decision-making. By integrating real-time biological data with adaptive computer algorithms, AI enhances therapeutic decision-making beyond the constraints of traditional static treatment models. This dynamic, data-driven model positions AI as the foundation of advanced precision immunotherapy for ocular malignancies.242,243
AI-based modeling of ocular immune dynamics
Conceptual frameworks suggest that cognitively adaptive immunotherapy, in which AI-assisted systems help regulate intraocular immune responses, represents a forward-looking and exploratory direction rather than a clinically established therapeutic option for ocular cancer. Conceptual frameworks have proposed the future integration of AI-driven immune modeling with localized ocular immunotherapy. These models envision real-time analysis of immune biomarkers, metabolic signals, and spatial immune dynamics to inform adaptive treatment strategies. These biocompatible graphene-based polymer devices feature sophisticated immune sensors that continuously monitor cytokine variations (e.g., IFN-γ, IL-10, TNF-α), detect immune cell localization via CD3/CD8/CD4 expression, and track metabolic indicators such as hypoxia-induced lactate accumulation in real time.244
Subsequent to data collection, the NSIPs evaluate these signals utilizing an integrated hybrid AI engine that amalgamates deep learning architectures with logic-based reasoning frameworks, representing a variant of neurosymbolic artificial intelligence. This system analyzes the immunological environment and determines context-specific therapeutic interventions.241 For instance, when T-cell fatigue markers like PD-1 or LAG-3 exceed baseline levels, the NSIP can administer anti-checkpoint chemicals through nanoporous micro-reservoirs or by activating optogenetic light pulses that rejuvenate local cytotoxic T-cells.245 Significantly, these treatments are not pre-coded; they are generated dynamically in real time by a distant, cloud-based computing system referred to as the Generative Immune Architecture (GIA).246 The GIA perpetually models and simulates tumor-immune interactions throughout time, subsequently relaying updated "immunologic scripts" to the NSIP, directing it on the timing and manner of immune activation or suppression.246 Together, these approaches illustrate a conceptual, device-centered closed-loop framework in which intraocular immune sensing and local actuation are dynamically coupled.241,245 Simultaneously, intelligent nanocarriers traversing the ocular vasculature are integrated with the identical AI network, facilitating accurate, condition-specific drug delivery.247 Immune-active substances like IL-2, bispecific T-cell engagers, or synthetic neoantigen constructions are released by these nano systems. Release is triggered only when specific biochemical signs are detected, such as hypoxia, TGF-β overexpression, or regulatory T-cell formation.245,247
Retinal optoelectronic implants with optogenetic immune switches enable light-directed immune modulation, wherein AI-controlled light frequencies selectively elicit pro-inflammatory or immunosuppressive responses according to the local immunological milieu.241,247 When combined, these ideas highlight the possibility of more contextually aware and spatially regulated immune regulation in the eye, although they are yet exploratory and need more experimental support. Such frameworks are conceptually described as adaptive systems that would continuously assess immune signals and inform context-dependent interventions, as opposed to fixed dosing paradigms.247,248 With sufficient experimental validation, such conceptual frameworks may help inform future strategies aimed at improving tumor control while preserving ocular immune balance.249 Although fully autonomous immune-regulating systems are not yet clinically available, incremental advances in biosensing, immuno-imaging, and adaptive drug delivery suggest that partial clinical translation of these concepts is feasible. This section introduces a number of AI-related ideas that are used as functional descriptors rather than as references to clinically deployed technologies.241 To be clear, "Generative Immune Architecture" refers to a hypothetical AI system that may dynamically simulate tumor-immune interactions and guide adaptive therapy choices by combining multi-omic, imaging, and clinical data.241 "Neural-symbolic immunologic processors" refer to hybrid computational systems that analyze immunological statuses and direct the choice of intervention by fusing rule-based immunologic logic with data-driven learning.247 Treatment paradigms where therapeutic parameters (such as timing, dosage, or target selection) are repeatedly modified based on real-time immune response are referred to as "cognitively adaptive immunotherapy".248 Similarly, "logic-gated antibodies" are created structures that improve spatial specificity by activating immune effector functions only when specified biological circumstances are satisfied at the same time.247,248 Lastly, rather than using set treatment regimens, "immunologic scripts" are a conceptual shorthand for algorithmically created decision rules that control context-dependent immune modulation.241,247,248
AI and computational immunology to optimize personalized immunotherapy
This section focuses on computational and machine-learning approaches that are already being used in modern cancer immunology to support patient stratification, biomarker discovery, and immunotherapy optimization, as opposed to the conceptual and device-oriented frameworks discussed in the section before.250 In this context, AI-based computational approaches are increasingly being applied to refine patient stratification, biomarker identification, and treatment optimization in cancer immunotherapy. The advancement of modern immunotherapy is increasingly reliant on machine learning algorithms that can simulate the complex interactions between malignancies and immune networks.250 Similar network-based modeling approaches developed in systems neuroscience, including harmonic and connectivity-aware frameworks, have demonstrated how complex biological interactions can be decomposed and analyzed at scale, providing transferable methodological insights for immune-network modeling.251 These computational methods are essential for forecasting therapy success, identifying probable responders, and mitigating immune-related toxicity. Elemam et al utilized 20 different machine learning methods to forecast immunotherapy responses in solid tumors, attaining very accurate survival forecasts via multi-omic data integration.250 Simultaneously, single-cell multi-omics technologies have facilitated AI-driven models to reveal immunological heterogeneity with unparalleled granularity and accuracy. Zhu et al combined machine learning algorithms with single-cell transcriptome datasets to delineate immune infiltration landscapes and guide the appropriate selection of checkpoint blockade medicines, thus enhancing the personalization of cancer immunotherapy.252 Next-generation CAR-T constructs are being computationally engineered to dynamically modify their activity in response to the tumor microenvironment, as proposed by Toro-Pedroza et al, representing a substantial advancement toward adaptive and self-regulating cellular immunotherapies.253 A nascent domain encompasses AI-assisted vaccine design, wherein computer tools facilitate the creation of highly tailored immunogens. Imani et al utilized deep learning frameworks to create neoantigen-based mRNA vaccines tailored to individual tumor characteristics, including pharmacokinetic aspects, in their computer modeling to improve vaccine efficacy and delivery accuracy.254 AI-engineered nanoparticle delivery systems have significantly improved focused therapeutic accuracy, facilitated tumor-specific administration, and reduced systemic toxicity. Simultaneously, artificial intelligence is transforming clinical decision support systems (CDSS) by offering oncologists personalized, data-informed treatment recommendations.254,255 Barua et al demonstrated this potential by illustrating how AI can derive actionable insights from unstructured electronic health records (EHRs) and scholarly literature.255 AI-driven reinforcement learning frameworks can now simulate immunotherapy trajectories to dynamically adjust dose schedules and treatment timing. Causal inference frameworks based on latent variable modeling, originally developed for analyzing neural spiking time-series, provide a conceptual foundation for disentangling dynamic immune interactions from noisy longitudinal immunologic data.256 Khanal et al have verified this unique technique, demonstrating its efficacy in neuroimmune modulation tests and emphasizing the potential of adaptive, self-learning algorithms to enhance and customize immunotherapy regimens in real time.257 While many precision immune engineering approaches have been put forward to treat ocular cancers, there is a significant disparity in the experimental validation of these approaches.139 Preclinical and early translational studies showing immune activation and antigen-specific targeting support dendritic cell-based vaccines and bispecific T-cell engagers, while AI-guided neoantigen prediction, adaptive dosing frameworks, and fully personalized immunogenomic platforms are currently mostly theoretical or computational. Different strategies have different benefits and drawbacks.139 Although vaccine-based strategies provide immunologic memory and specificity, they rely on efficient antigen presentation, which is often impaired in ocular malignancies. Bispecific designs have the potential to cause off-target inflammation yet may directly engage effector T cells to circumvent inadequate priming.146 Delivery methods based on nanocarriers increase local bioavailability and lower systemic toxicity, but they have issues with regulated release, ocular penetration, and retention.139,146 Crucially, delivery limitations specific to ocular anatomy, such as blood–retinal barrier impermeability, small injection volumes, and the need to maintain visual function, must be addressed by immunological engineering techniques in the eye.146 These elements highlight the need for precise, localized delivery methods and warn against applying systemic immunotherapy paradigms directly to ocular settings.
Technical and translational limitations of AI-powered ocular immunotherapy
The practical usefulness of AI-enabled implants, intelligent nanocarriers, and optogenetic immune-modulation platforms is still limited by significant translational and technological obstacles, despite their conceptual attractiveness.258 Strict anatomical restrictions, restricted surgical access, and the possibility of vision-threatening side effects, including infection, retinal detachment, or chronic inflammation, must all be considered when implantation of sensor or actuation devices into the eye.259 Because persistent foreign elements might eventually cause fibrotic encapsulation, immunological activation, or device failure, long-term biocompatibility is still a major problem.260 From an immunological standpoint, autoimmune uveitis, off-target inflammation, and permanent loss of ocular immune privilege are risks associated with frequent or prolonged immune manipulation inside the eye.261 Although localized immune activation has potential benefits, a major obstacle is still obtaining enough therapeutic efficiency without causing collateral tissue harm.261 Furthermore, there are further complications associated with maintaining implanted or semi-autonomous systems, such as the necessity for modification or removal operations, calibration drift, signal stability, and device endurance.262 Lastly, the majority of AI-guided immunotherapeutic platforms still rely on upstream developments in real-time immunologic monitoring, biosensing accuracy, and regulatory frameworks that can assess self-modifying or adaptive medical systems.263 Therefore, rather than being immediate therapeutic remedies, these technologies should now be seen as long-term translational goals.264 Iterative preclinical validation, cautious clinical trial design, and tight coordination across immunology, materials science, and surgical innovation are all necessary for further advancement.262-264
Ethical and practical considerations: trial design in rare ocular tumors
Clinical trials with rare eye cancers, such as uveal melanoma, conjunctival carcinoma, retinoblastoma, and ocular adnexal lymphomas, present unique ethical and practical obstacles. Despite representing a minuscule percentage of worldwide cancer incidence, these malignancies significantly affect vision and survival, hence requiring highly personalized, ethically sound research approaches that harmonize scientific integrity with patient safety and quality of life factors.
Ethical challenges
The idea of clinical equipoise, which requires authentic ambiguity about the superiority of one treatment compared to another, is sometimes contested in rare tumor trials due to the scarcity of comparison data. Ethical decision-making becomes more complex when standard therapies provide limited benefit and experimental options are scarce. In these situations, denying access to potentially advantageous investigational medicines, even as a control measure, may contravene the ethical principle of beneficence. Furthermore, informed consent in these investigations necessitates extraordinary rigor. Individuals with uncommon ocular malignancies frequently exhibit "therapeutic optimism," mistakenly associating participation in clinical trials with assured therapeutic advantage. Consequently, the consent procedure must be meticulously designed to convey uncertainty, potential hazards, and realistic outcomes with complete transparency. The ethical considerations grow further intricate in pediatric instances, such as retinoblastoma, when proxy consent by guardians creates further moral and legal issues that necessitate meticulous management.
Practical and methodological constraints
Conventional randomized controlled trials (RCTs) are frequently unfeasible for uncommon ocular malignancies due to very restricted patient groups. As a result, other trial designs have arisen as ethical and statistically valid possibilities. Adaptive trial designs, Bayesian statistical frameworks, and basket trial methodologies provide variable patient recruitment and iterative data-informed learning as evidence is gathered. Pathak and Singh assert that Bayesian approaches, namely, achieve a judicious equilibrium between statistical accuracy and practical applicability in the context of small patient populations. N-of-1 trial designs, although infrequently utilized, offer ethically advantageous frameworks, particularly for ultra-rare ocular cancers. These trials provide highly personalized therapy assessment while ensuring scientific rigor and upholding ethical transparency in patient-centered research.
Global collaboration and data pooling
International collaboration is increasingly necessary to address limited patient numbers and support data integration in rare ocular tumors. Initiatives like the European Reference Network for Rare Eye Diseases (ERN-EYE) and the International Rare Cancers Initiative (IRCI) are crucial for standardizing research techniques, facilitating data exchange, and allowing meta-analytic integration among institutions. Nonetheless, these collaborative initiatives present considerable hurdles, such as complexities in cross-border ethical reviews, data privacy issues, compliance with GDPR requirements, and the necessity of guaranteeing equitable access to research participation and new medicines for all patients engaged.
Justice and accessibility
Ensuring equal access to clinical trials for underrepresented populations, especially in resource-constrained areas, constitutes a core ethical responsibility in contemporary ocular oncology research. To reduce equity disparities and enhance geographic inclusion, innovative approaches such as telemedicine platforms, decentralized patient recruitment models, and AI-driven screening systems are being utilized, facilitating remote participation, prompt diagnosis, and more inclusive trial enrollment among varied populations (Supplementary file 1,Table S1).
Discussion
Across current immunotherapy platforms for uveal melanoma, a recurring pattern is observed. Only certain facets of ocular immune privilege are addressed by each modality, and no one strategy has been shown to be consistently effective.6 TIL-, CAR-T-, and TCR-T–based therapies are frequently limited by metabolic suppression and early T-cell exhaustion; oncolytic viruses can induce inflammation but remain constrained by delivery challenges; bispecific TCR therapies produce more consistent responses yet are restricted by HLA subtype and microenvironmental resistance; checkpoint inhibitors fail primarily due to insufficient inflammatory priming; and vaccine-based strategies often induce systemic immunity without effective tumor infiltration.268 These observations indicate that resistance to checkpoint inhibition in uveal melanoma arises not from intrinsic drug failure, but from a convergence of immune privilege, low neoantigen burden, impaired antigen presentation, and dominant local immunosuppressive signals.268 As a result, checkpoint inhibitors alone cannot overcome the structural and functional barriers that define the ocular tumor microenvironment.268 Accordingly, uveal melanoma immunotherapy may be better understood as a problem of coordinated immune modulation rather than the optimization of a single therapeutic modality.93 Effective treatment is therefore likely to require combination strategies that sequentially promote tumor priming, enhance effector-cell engagement, and limit suppressive metabolic and cytokine pathways such as IDO or TGF-β.143 Immune privilege can be conceptualized as a dynamically regulated biological system that may be transiently modified and subsequently restored to preserve ocular integrity. These insights support the development of rational combination approaches that integrate immune activation, trafficking, and metabolic support in a coordinated manner.269
Research restrictions and unresolved issues
The therapeutic effect of immune-based treatments for ocular cancers is still limited by a number of outstanding issues, despite significant advancements in this field.6 The most important of them is the eye's immune-privileged condition, which places strict restrictions on immune-cell trafficking, inflammatory amplification, and therapeutic penetration.93 This privilege is necessary to maintain visual function, but it also complicates the direct application of systemic immunotherapy paradigms by fostering a milieu that prioritizes immune tolerance over long-lasting anticancer immunity.270 The molecular features of ocular malignancies themselves constitute a similar constraint.270 The most notable characteristics of uveal melanoma are low tumor mutational load, limited neoantigen variety, and inadequate antigen presentation, which lead to poor baseline T-cell priming.60 These characteristics essentially limit checkpoint inhibition's effectiveness and contribute to the explanation of the low response rates seen in clinical studies, especially when combination tactics are used.189 Technical and practical obstacles sometimes hinder advancement. Because of the possibility of vision-threatening consequences, intraocular tumor biopsies are often avoided, which restricts access to longitudinal tissue-based immune profiling.271 While new methods like liquid biopsy, imaging biomarkers, and spatial transcriptomics provide useful surrogate data, they are not yet able to completely replace direct tumor–immune interface investigation.194 Another limitation is safety issues. The therapeutic window for effective immunological treatments is limited because excessive immune activation in the small ocular region might result in autoimmune uveitis, retinal damage, and permanent vision impairment.194 Instead of long-term systemic activation, this calls for finely regulated, targeted, and reversible immune modulation techniques. When taken as a whole, these restrictions suggest that no one immunotherapeutic approach is likely to be adequate for treating ocular cancers.271 Rather, sensible combination strategies that include immune priming, microenvironmental reprogramming, and precise administration while honoring the anatomical and functional limitations specific to the eye will be necessary for successful clinical translation.190
Conclusion
Ocular cancers, especially uveal melanoma, demonstrate the basic incompatibility between immune-privileged, anatomically limited tissues and systemic immunotherapy models. Immune checkpoint inhibition is constrained by low tumor mutational burden, poor antigen presentation, inadequate baseline T-cell priming, and dominant local immunosuppressive networks maintained by ocular immune privilege. This biological pattern is consistent across all currently available platforms. Together, these characteristics account for the mild and sometimes temporary clinical improvements seen in ocular malignancies when checkpoint blockade is used alone. The inability to safely and reversibly modulate immune privilege without compromising vision, restricted access to longitudinal tumor tissue for immune monitoring, a limited therapeutic window imposed by the risk of autoimmune uveitis, and insufficient delivery strategies capable of limiting immune activation to tumor-relevant compartments are some of the major unmet needs identified in this review that continue to impede therapeutic progress. Approaches that combine biological effectiveness with spatial accuracy and ocular safety are needed to address these issues. The greatest practical short-term progress, from a translational perspective, is found in approaches that specifically tackle these obstacles. These include localized and biomaterial-enabled drug delivery systems that limit immune modulation to intraocular or peri-tumoral spaces, TCR-based redirection platforms that can avoid defective antigen presentation, targeted suppression of metabolic and cytokine checkpoints like IDO and TGF-β, and adaptive clinical trial designs that are adapted to the rarity and heterogeneity of ocular tumors. These strategies provide tenable avenues for deployment and are backed by newly available clinical or late-preclinical data. Conversely, more futuristic ideas, like as implanted immune-responsive devices, completely autonomous AI-guided immune control systems, and optogenetic immunity switches, need to be seen as long-term translational goals rather than short-term treatment fixes. Advances in biosensing dependability, long-term biocompatibility, regulatory supervision for adaptive medical systems, and thorough safety validation are necessary for their implementation. Together, our findings support a precision immune engineering paradigm wherein logical combination techniques, spatially controlled administration, and reversible immune privilege modulation, rather than systemic immune activation, drive advances in ocular immunotherapy. While AI-driven closed-loop immune regulation is still primarily theoretical and will necessitate rigorous biomarker validation and delivery strategies specifically tailored to ocular safety constraints, ocular immuno-oncology 2.0 represents a translational continuum in which therapeutically viable approaches are starting to emerge.
Review Highlights
What is the current knowledge?
-
Ocular immune privilege restricts immune-cell trafficking and limits systemic immunotherapy efficacy.
-
Immune checkpoint inhibitors show low and short-lived responses in uveal melanoma.
-
Tumor immune resistance arises from low antigenicity and dominant local immunosuppressive pathways.
What is new here?
-
This review reframes ocular immunotherapy as a problem of spatially constrained immune engineering.
-
It integrates delivery systems, immune redirection, and metabolic modulation as coordinated solutions.
-
Translational priorities are identified, distinguishing near-term strategies from long-term conceptual technologies.
Competing Interests
The authors declare that they have no competing interests related to this work.
Data Availability Statement
No new data were generated or analyzed during this study.
Ethical Approval
This article is a narrative review and does not contain any studies involving human participants or animals performed by the authors. No personal data is included in this manuscript.
Declaration of AI-assisted Tools in the Writing Procedure
AI-assisted tools (ChatGPT) were used exclusively for language refinement and clarity improvement. All scientific content, interpretations, and conclusions were developed by the authors.
Supplementary files
Supplementary file 1 contains Table S1.
(pdf)
Acknowledgements
Figures were created with BioRender.com under an institutional license.
References
- Chen K-Y, Chan H-C, Chan C-M. Can Immune Checkpoint Modulation Redefine Ocular Immunotherapy? Emerging Mechanisms, Challenges, and Translational Opportunities-A Comprehensive Review. Invest Ophthalmol Vis Sci 2025; 66:53. doi: 10.1167/iovs.66.14.53 [Crossref] [ Google Scholar]
- Reddy R V, Ong J, Lee R, Sampige R, Waisberg E, Gibson CR. Space radiation and risk for ocular surface malignancies: Exposure risk, current mitigation strategies, and management considerations for a mission to Mars. Life Sci Sp Res 2025; 47:69-76. doi: 10.1016/j.lssr.2025.06.002 [Crossref] [ Google Scholar]
- Ma S, Huis In’t Veld R V, Pinos E de L, Ossendorp FA, Jager MJ. Targeting ocular malignancies using a novel light-activated virus-like drug conjugate. Adv OphthalmolPract Res 2025; 5:49-57. doi: 10.1016/j.aopr.2024.12.001 [Crossref] [ Google Scholar]
- Cho JH, Nam SY, Jo J. High-density Lipoprotein Cholesterol and the Development of Extranodal Marginal Zone B-cell Lymphoma of Mucosa-associated Lymphoid Tissue. J Cancer Prev 2025; 30:89-96. doi: 10.15430/JCP.25.006 [Crossref] [ Google Scholar]
- He Y, Gao C. Risk and clinical implications of histological transformation in extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue: A population-based analysis. Sci Rep 2025; 15:20407. doi: 10.1038/s41598-025-07413-8 [Crossref] [ Google Scholar]
-
Song J, Mou P, Song G-G, Chen L, Chen Y-Q, Wei R-L. Advances in immunotherapy for uveal melanoma: enhancing efficacy and overcoming resistance. Front Cell Dev Biol 2025;13:1619150. doi: 10.3389/fcell.2025.1619150.
- Zhang X, You W, Wang Y, Dejenie R, Wang C, Huang Y. Prospects of anti-GD2 immunotherapy for retinoblastoma. Front Immunol 2024; 15:1499700. doi: 10.3389/fimmu.2024.1499700 [Crossref] [ Google Scholar]
- Yang Y, Jin X, Xu M, He W. Clinical characteristics and treatment outcomes of patients with IgG4-positive ocular adnexal marginal zone B-cell lymphoma. Indian J Ophthalmol 2024; 72:S902-S906. doi: 10.4103/IJO.IJO_2560_23 [Crossref] [ Google Scholar]
- Tan Y, Lu Y, Chen S, Zou C, Qin B. Immunotherapy for ocular melanoma: a bibliometric and visualization analysis from 1991 to 2022. Front Oncol 2023; 13:1161759. doi: 10.3389/fonc.2023.1161759 [Crossref] [ Google Scholar]
- Ziogas DC, Foteinou D, Theocharopoulos C, Martinos A, Petsiou D-P, Anastasopoulou A. State-of-the-art in Metastatic Uveal Melanoma Treatment: A 2025 Update. Curr Oncol Rep 2025; 27:803-821. doi: 10.1007/s11912-025-01684-0 [Crossref] [ Google Scholar]
- Yamada K, Takeuchi M, Fukumoto T, Suzuki M, Kato A, Mizuki Y. Immune checkpoint inhibitors for metastatic uveal melanoma: a meta-analysis. Sci Rep 2024; 14:7887. doi: 10.1038/s41598-024-55675-5 [Crossref] [ Google Scholar]
- Wang J, Li Z, Yin H. The Future of Checkpoint Inhibitors in Uveal Melanoma: A Narrative Review. OphthalmolTher 2024; 13:1103-1123. doi: 10.1007/s40123-024-00913-2 [Crossref] [ Google Scholar]
- Virgili G, Gatta G, Ciccolallo L, Capocaccia R, Biggeri A, Crocetti E. Incidence of uveal melanoma in Europe. Ophthalmology 2007; 114:2309-2315. doi: 10.1016/j.ophtha.2007.01.032 [Crossref] [ Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N Engl J Med 2010; 363:711-723. doi: 10.1056/NEJMoa1003466 [Crossref] [ Google Scholar]
- Rozeman EA, Prevoo W, Meier MAJ, Sikorska K, Van TM, van de Wiel BA. Phase Ib/II trial testing combined radiofrequency ablation and ipilimumab in uveal melanoma (SECIRA-UM). Melanoma Res 2020; 30:252-260. doi: 10.1097/CMR.0000000000000653 [Crossref] [ Google Scholar]
- Bol KF, Ellebaek E, Hoejberg L, Bagger MM, Larsen MS, Klausen TW. Real-world impact of immune checkpoint inhibitors in metastatic uveal melanoma. Cancers (Basel) 2019; 11:1489. doi: 10.3390/cancers11101489 [Crossref] [ Google Scholar]
- Khoja L, Atenafu EG, Suciu S, Leyvraz S, Sato T, Marshall E. Meta-analysis in metastatic uveal melanoma to determine progression free and overall survival benchmarks: an international rare cancers initiative (IRCI) ocular melanoma study. Ann Oncol 2019; 30:1370-1380. doi: 10.1093/annonc/mdz176 [Crossref] [ Google Scholar]
- Koch EAT, Petzold A, Wessely A, Dippel E, Gesierich A, Gutzmer R. Immune checkpoint blockade for metastatic uveal melanoma: patterns of response and survival according to the presence of hepatic and extrahepatic metastasis. Cancers (Basel) 2021; 13:3359. doi: 10.3390/cancers13133359 [Crossref] [ Google Scholar]
- Joshua AM, Monzon JG, Mihalcioiu C, Hogg D, Smylie M, Cheng T. A phase 2 study of tremelimumab in patients with advanced uveal melanoma. Melanoma Res 2015; 25:342-347. doi: 10.1097/CMR.0000000000000175 [Crossref] [ Google Scholar]
- Diener-West M, Reynolds SM, Agugliaro DJ, Caldwell R, Cumming K, Earle JD, et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch Ophthalmol2005.123:1639–1643. doi: 10.1001/archopht.123.12.1639.
- Wang JZ, Lin V, Toumi E, Wang K, Zhu H, Conway RM. Development of new therapeutic options for the treatment of uveal melanoma. FEBS J 2021; 288:6226-6249. doi: 10.1111/febs.15869 [Crossref] [ Google Scholar]
- Singh L, Singh MK, Kenney MC, Jager MJ, Rizvi MA, Meel R. Prognostic significance of PD-1/PD-L1 expression in uveal melanoma: correlation with tumor-infiltrating lymphocytes and clinicopathological parameters. Cancer Immunol Immunother 2021; 70:1291-1303. doi: 10.3390/cancers13164031 [Crossref] [ Google Scholar]
- Robert C, Long G V, Brady B, Dutriaux C, Maio M, Mortier L. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320-330. doi: 10.1056/NEJMoa1412082 [Crossref] [ Google Scholar]
- Namikawa K, Takahashi A, Mori T, Tsutsumida A, Suzuki S, Motoi N. Nivolumab for patients with metastatic uveal melanoma previously untreated with ipilimumab: a single-institution retrospective study. Melanoma Res 2020; 30:76-84. doi: 10.1097/CMR.0000000000000617 [Crossref] [ Google Scholar]
- Tacar SY, Selcukbiricik F, Yilmaz M, Erturk K, Sarici AM, Gulturk I. Nivolumab for metastatic uveal melanoma: a multicenter, retrospective study. Melanoma Res 2021; 31:449-455. doi: 10.1097/CMR.0000000000000744 [Crossref] [ Google Scholar]
- Karydis I, Chan PY, Wheater M, Arriola E, Szlosarek PW, Ottensmeier CH. Clinical activity and safety of Pembrolizumab in Ipilimumab pre-treated patients with uveal melanoma. Oncoimmunology 2016; 5:e1143997. doi: 10.1080/2162402X.2016.1143997 [Crossref] [ Google Scholar]
- Kottschade LA, McWilliams RR, Markovic SN, Block MS, Bisneto JV, Pham AQ. The use of pembrolizumab for the treatment of metastatic uveal melanoma. Melanoma Res 2016; 26:300-303. doi: 10.1097/CMR.0000000000000242 [Crossref] [ Google Scholar]
- Johnson DB, Bao R, Ancell KK, Daniels AB, Wallace D, Sosman JA. Response to anti–PD-1 in uveal melanoma without high-volume liver metastasis. J Natl Compr Cancer Netw 2019; 17:114-117. doi: 10.6004/jnccn.2018.7070 [Crossref] [ Google Scholar]
- Rossi E, Pagliara MM, Orteschi D, Dosa T, Sammarco MG, Caputo CG. Pembrolizumab as first-line treatment for metastatic uveal melanoma. Cancer Immunol Immunother 2019; 68:1179-1185. doi: 10.1007/s00262-019-02352-6 [Crossref] [ Google Scholar]
- Jansen YJL, Seremet T, Neyns B. Pembrolizumab for the treatment of uveal melanoma: a case series. Rare Tumors 2020; 12:2036361320971983. doi: 10.1177/2036361320971983 [Crossref] [ Google Scholar]
- Algazi AP, Tsai KK, Shoushtari AN, Munhoz RR, Eroglu Z, Piulats JM. Clinical outcomes in metastatic uveal melanoma treated with PD‐1 and PD‐L1 antibodies. Cancer 2016; 122:3344-3353. doi: 10.1002/cncr.30258 [Crossref] [ Google Scholar]
- Heppt M V, Heinzerling L, Kähler KC, Forschner A, Kirchberger MC, Loquai C. Prognostic factors and outcomes in metastatic uveal melanoma treated with programmed cell death-1 or combined PD-1/cytotoxic T-lymphocyte antigen-4 inhibition. Eur J Cancer 2017; 82:56-65. doi: 10.1016/j.ejca.2017.05.038 [Crossref] [ Google Scholar]
- Bender C, Enk A, Gutzmer R, Hassel JC. Anti‐PD‐1 antibodies in metastatic uveal melanoma: a treatment option?. Cancer Med 2017; 6:1581-1586. doi: 10.1002/cam4.887 [Crossref] [ Google Scholar]
- Pelster MS, Gruschkus SK, Bassett R, Gombos DS, Shephard M, Posada L. Nivolumab and Ipilimumab in Metastatic Uveal Melanoma: Results From a Single-Arm Phase II Study. J Clin Oncol 2021; 39:599-607. doi: 10.1200/JCO.20.00605 [Crossref] [ Google Scholar]
- Nathan P, Hassel JC, Rutkowski P, Baurain J-F, Butler MO, Schlaak M. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N Engl J Med 2021; 385:1196-1206. doi: 10.1056/NEJMoa2103485 [Crossref] [ Google Scholar]
- Kato J, Uhara H. Immunotherapy for advanced melanoma: current situation in Japan. Jpn J Clin Oncol 2020; 51:3-9. doi: 10.1093/jjco/hyaa188 [Crossref] [ Google Scholar]
- Piulats JM, Espinosa E, de la Cruz Merino L, Varela M, Alonso Carrión L, Martín-Algarra S. Nivolumab Plus Ipilimumab for Treatment-Naïve Metastatic Uveal Melanoma: An Open-Label, Multicenter, Phase II Trial by the Spanish Multidisciplinary Melanoma Group (GEM-1402). J Clin Oncol 2021; 39:586-598. doi: 10.1200/JCO.20.00550 [Crossref] [ Google Scholar]
- Heppt M V, Amaral T, Kähler KC, Heinzerling L, Hassel JC, Meissner M. Combined immune checkpoint blockade for metastatic uveal melanoma: a retrospective, multi-center study. J Immunother Cancer 2019; 7:299. doi: 10.1186/s40425-019-0800-0 [Crossref] [ Google Scholar]
-
Najjar YG, Navrazhina K, Ding F, Bhatia R, Tsai K, Abbate K, et al. Ipilimumab plus nivolumab for patients with metastatic uveal melanoma: a multicenter, retrospective study. J Immunother Cancer 2020;8:e000331. doi: 10.1136/jitc-2019-000331.
- Karivedu V, Eldessouki I, Taftaf A, Zhu Z, Makramalla A, Karim NA. Nivolumab and Ipilimumab in the Treatment of Metastatic Uveal Melanoma: A Single-Center Experience. Case Rep Oncol Med 2019; 2019:3560640. doi: 10.1155/2019/3560640 [Crossref] [ Google Scholar]
- Vergara IA, Wilmott JS, Long G V, Scolyer RA. Genetic drivers of non-cutaneous melanomas: Challenges and opportunities in a heterogeneous landscape. Exp Dermatol 2022; 31:13-30. doi: 10.1111/exd.14287 [Crossref] [ Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin A V. Signatures of mutational processes in human cancer. Nature 2013; 500:415-421. doi: 10.1038/nature12477 [Crossref] [ Google Scholar]
- Basile MS, Mazzon E, Fagone P, Longo A, Russo A, Fallico M. Immunobiology of Uveal Melanoma: State of the Art and Therapeutic Targets. Front Oncol 2019; 9:1145. doi: 10.3389/fonc.2019.01145 [Crossref] [ Google Scholar]
- Qin Y, Petaccia de Macedo M, Reuben A, Forget M-A, Haymaker C, Bernatchez C. Parallel profiling of immune infiltrate subsets in uveal melanoma versus cutaneous melanoma unveils similarities and differences: A pilot study. Oncoimmunology 2017; 6:e1321187. doi: 10.1080/2162402X.2017.1321187 [Crossref] [ Google Scholar]
- Durante MA, Rodriguez DA, Kurtenbach S, Kuznetsov JN, Sanchez MI, Decatur CL. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat Commun 2020; 11:496. doi: 10.1038/s41467-019-14256-1 [Crossref] [ Google Scholar]
- Seth R, Messersmith H, Kaur V, Kirkwood JM, Kudchadkar R, McQuade JL. Systemic Therapy for Melanoma: ASCO Guideline. J Clin Oncol 2020; 38:3947-3970. doi: 10.1200/JCO.20.00198 [Crossref] [ Google Scholar]
- Rao PK, Barker C, Coit DG, Joseph RW, Materin M, Rengan R. NCCN Guidelines Insights: Uveal Melanoma, Version 12019: Featured Updates to the NCCN Guidelines. J Natl Compr Cancer Netw J Natl ComprCancNetw 2020; 18:120-131. doi: 10.6004/jnccn.2020.0007 [Crossref] [ Google Scholar]
- Harjunpää H, Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol 2020; 200:108-119. doi: 10.1111/cei.13407 [Crossref] [ Google Scholar]
- Chauvin J-M, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J Clin Invest 2015; 125:2046-2058. doi: 10.1172/JCI80445 [Crossref] [ Google Scholar]
- Stålhammar G, Seregard S, Grossniklaus HE. Expression of immune checkpoint receptors Indoleamine 2,3-dioxygenase and T cell Ig and ITIM domain in metastatic versus nonmetastatic choroidal melanoma. Cancer Med 2019; 8:2784-2792. doi: 10.1002/cam4.2167 [Crossref] [ Google Scholar]
- Masaoutis C, Kokkali S, Theocharis S. Immunotherapy in uveal melanoma: novel strategies and opportunities for personalized treatment. Expert OpinInvestig Drugs 2021; 30:555-569. doi: 10.1080/13543784.2021.1898587 [Crossref] [ Google Scholar]
- Zhai L, Ladomersky E, Lenzen A, Nguyen B, Patel R, Lauing KL. IDO1 in cancer: a Gemini of immune checkpoints. Cell Mol Immunol 2018; 15:447-457. doi: 10.1038/cmi.2017.143 [Crossref] [ Google Scholar]
- Rossi E, Schinzari G, Zizzari IG, Maiorano BA, Pagliara MM, Sammarco MG. Immunological Backbone of Uveal Melanoma: Is There a Rationale for Immunotherapy?. Cancers (Basel) 2019; 11:1055. doi: 10.3390/cancers11081055 [Crossref] [ Google Scholar]
- Mitchell TC, Hamid O, Smith DC, Bauer TM, Wasser JS, Olszanski AJ. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J Clin Oncol 2018; 36:3223-3230. doi: 10.1200/JCO.2018.78.9602 [Crossref] [ Google Scholar]
- Long G V, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol 2019; 20:1083-1097. doi: 10.1016/S1470-2045(19)30274-8 [Crossref] [ Google Scholar]
- Li Y-Z, Huang Y, Deng X-Y, Tu C-S. Identification of an immune-related signature for the prognosis of uveal melanoma. Int J Ophthalmol 2020; 13:458-465. doi: 10.18240/ijo.2020.03.14 [Crossref] [ Google Scholar]
- Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med 1990; 171:1393-1405. doi: 10.1084/jem.171.5.1393 [Crossref] [ Google Scholar]
- Souri Z, Wierenga APA, Kroes WGM, van der Velden PA, Verdijk RM, Eikmans M. LAG3 and its ligands show increased expression in high-risk uveal melanoma. Cancers (Basel) 2021; 13:4445. doi: 10.3390/cancers13174445 [Crossref] [ Google Scholar]
- Woo S-R, Turnis ME, Goldberg M V, Bankoti J, Selby M, Nirschl CJ. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res 2012; 72:917-927. doi: 10.1158/0008-5472.CAN-11-1620 [Crossref] [ Google Scholar]
- Li WJ, Najdawi W, Badla O, Galor A, Karp CL. Immune Checkpoint Inhibitors in the Treatment of Ocular Surface Cancers: A Review. Semin Ophthalmol 2025; 40:485-495. doi: 10.1080/08820538.2025.2458658 [Crossref] [ Google Scholar]
- Gong Y, Liu Y, Jiang F, Wang X. Ocular Immune-Related Adverse Events Associated with PD-1 Inhibitors: From Molecular Mechanisms to Clinical Management. Semin Ophthalmol 2025; 40:288-305. doi: 10.1080/08820538.2024.2433636 [Crossref] [ Google Scholar]
- Zhao L, Xia W, Zhang Y, Zou P, Zhu Q, Zhang R. Efficacy and Safety of Immune Checkpoint Blockades in the Treatment of Ocular Melanoma: A Systematic Review and Meta-Analysis. Front Oncol 2021; 11:781162. doi: 10.3389/fonc.2021.781162 [Crossref] [ Google Scholar]
- Wen J, Mao X, Cheng Q, Liu Z, Liu F. A pan-cancer analysis revealing the role of TIGIT in tumor microenvironment. Sci Rep 2021; 11:22502. doi: 10.1038/s41598-021-01933-9 [Crossref] [ Google Scholar]
- Parikh RA, Chaon BC, Berkenstock MK. Ocular Complications of Checkpoint Inhibitors and Immunotherapeutic Agents: A Case Series. Ocul Immunol Inflamm 2021; 29:1585-1590. doi: 10.1080/09273948.2020.1766082 [Crossref] [ Google Scholar]
- Adashek JJ, Kato S, Nishizaki D, Miyashita H, De P, Lee S. LAG-3 transcriptomic expression patterns across malignancies: Implications for precision immunotherapeutics. Cancer Med 2023; 12:13155-13166. doi: 10.1002/cam4.6000 [Crossref] [ Google Scholar]
- Bosch JJ, Iheagwara UK, Reid S, Srivastava MK, Wolf J, Lotem M. Uveal melanoma cell-based vaccines express MHC II molecules that traffic via the endocytic and secretory pathways and activate CD8+ cytotoxic, tumor-specific T cells. Cancer Immunol Immunother 2010; 59:103-112. doi: 10.1007/s00262-009-0729-0 [Crossref] [ Google Scholar]
- Dissanayake SK, Thompson JA, Bosch JJ, Clements VK, Chen PW, Ksander BR. Activation of Tumor-specific CD4+ T Lymphocytes by Major Histocompatibility Complex Class II Tumor Cell Vaccines: A Novel Cell-based Immunotherapy. Cancer Res 2004; 64:1867-1874. doi: 10.1158/0008-5472.CAN-03-2634 [Crossref] [ Google Scholar]
- Bosch JJ, Thompson JA, Srivastava MK, Iheagwara UK, Murray TG, Lotem M. MHC Class II–Transduced Tumor Cells Originating in the Immune-Privileged Eye Prime and Boost CD4+ T Lymphocytes that Cross-react with Primary and Metastatic Uveal Melanoma Cells. Cancer Res 2007; 67:4499-4506. doi: 10.1158/0008-5472.CAN-06-3770 [Crossref] [ Google Scholar]
- Bennett SRM, Carbone FR, Karamalis F, Miller JFAP, Heath WR. Induction of a CD8+ Cytotoxic T Lymphocyte Response by Cross-priming Requires Cognate CD4+ T Cell Help. J Exp Med 1997; 186:65-70. doi: 10.1084/jem.186.1.65 [Crossref] [ Google Scholar]
- J-A K, J F. Helper activity is required for the in vivo generation of cytotoxic T lymphocytes. J Exp Med 1982; 155:768-782. doi: 10.1084/jem.155.3.768 [Crossref] [ Google Scholar]
- Bennett SRM, Carbone FR, Karamalis F, Flavell RA, Miller JFAP, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998; 393:478-480. doi: 10.1038/30996 [Crossref] [ Google Scholar]
- Schoenberger SP, Toes REM, van der Voort EIH, Offringa R, Melief CJM. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature 1998; 393:480-483. doi: 10.1038/31002 [Crossref] [ Google Scholar]
- Takeuchi A, Saito T. CD4 CTL, a Cytotoxic Subset of CD4+ T Cells, Their Differentiation and Function. Front Immunol 2017; 8:194. doi: 10.3389/fimmu.2017.00194 [Crossref] [ Google Scholar]
- Schattner EJ, Mascarenhas J, Bishop J, Yoo DH, Chadburn A, Crow MK. CD4+ T-cell induction of Fas-mediated apoptosis in Burkitt’s lymphoma B cells. Blood 1996; 88:1375-1382. doi: 10.1182/blood.V88.4.1375.bloodjournal8841375 [Crossref] [ Google Scholar]
- Kittler JM, Sommer J, Fischer A, Britting S, Karg MM, Bock B. Characterization of CD4+ T cells primed and boosted by MHCII primary uveal melanoma cell-based vaccines. Oncotarget 2019; 10:1812-1828. doi: 10.18632/oncotarget.26737 [Crossref] [ Google Scholar]
- Schank TE, Hassel JC. Immunotherapies for the Treatment of Uveal Melanoma—History and Future. Cancers (Basel) 2019; 11:1048. doi: 10.3390/cancers11081048 [Crossref] [ Google Scholar]
- de Vries TJ, Trančikova D, Ruiter DJ, van Muijen GNP. High expression of immunotherapy candidate proteins gp100, MART-1, tyrosinase and TRP-1 in uveal melanoma. Br J Cancer 1998; 78:1156-1161. doi: 10.1038/bjc.1998.646 [Crossref] [ Google Scholar]
- Steuhl K-P, Rohrbach JM, Knorr M, Thiel H-J. Significance, Specificity, and Ultrastructural Localization of HMB-45 Antigen in Pigmented Ocular Tumors. Ophthalmology 1993; 100:208-215. doi: 10.1016/S0161-6420(93)31668-4 [Crossref] [ Google Scholar]
- Bol KF, van den Bosch T, Schreibelt G, Mensink HW, Keunen JEE, Kiliç E. Adjuvant Dendritic Cell Vaccination in High-Risk Uveal Melanoma. Ophthalmology 2016; 123:2265-2267. doi: 10.1016/j.ophtha.2016.06.027 [Crossref] [ Google Scholar]
- Rantala ES, Hernberg M, Kivelä TT. Overall survival after treatment for metastatic uveal melanoma: a systematic review and meta-analysis. Melanoma Res 2019; 29:561-8. doi: 10.1097/CMR.0000000000000575 [Crossref] [ Google Scholar]
- Chandran SS, Somerville RPT, Yang JC, Sherry RM, Klebanoff CA, Goff SL. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: a single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol 2017; 18:792-802. doi: 10.1016/S1470-2045(17)30251-6 [Crossref] [ Google Scholar]
- Hutchinson L. Cul-TIL-vating uveal melanoma regression. Nat Rev Clin Oncol 2017; 14:328-329. doi: 10.1038/nrclinonc.2017.61 [Crossref] [ Google Scholar]
- Gezgin G, Luk SJ, Cao J, Dogrusöz M, van der Steen DM, Hagedoorn RS. PRAME as a Potential Target for Immunotherapy in Metastatic Uveal Melanoma. JAMA Ophthalmol 2017; 135:541-549. doi: 10.1001/jamaophthalmol.2017.0729 [Crossref] [ Google Scholar]
- Al-Khadairi G, Decock J. Cancer Testis Antigens and Immunotherapy: Where Do We Stand in the Targeting of PRAME?. Cancers (Basel) 2019; 11:984. doi: 10.3390/cancers11070984 [Crossref] [ Google Scholar]
- Park J, Talukder AH, Lim SA, Kim K, Pan K, Melendez B. SLC45A2: A Melanoma Antigen with High Tumor Selectivity and Reduced Potential for Autoimmune Toxicity. Cancer Immunol Res 2017; 5:618-629. doi: 10.1158/2326-6066.CIR-17-0051 [Crossref] [ Google Scholar]
- Hofmann O, Caballero OL, Stevenson BJ, Chen Y-T, Cohen T, Chua R. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci 2008; 105:20422-20427. doi: 10.1073/pnas.0810777105 [Crossref] [ Google Scholar]
- Christophe L, Bernard L, Etienne DP, Véronique C, Ivan T, van Baren Nicolas. Contrasting frequencies of antitumor and anti-vaccine T cells in metastases of a melanoma patient vaccinated with a MAGE tumor antigen. J Exp Med 2005; 201:249-257. doi: 10.1084/jem.20041378 [Crossref] [ Google Scholar]
- Chodnicki KD, Prasad S. Ophthalmic Implications of Chimeric Antigen Receptor T-Cell Therapy. Semin Ophthalmol 2021; 36:329-334. doi: 10.1080/08820538.2021.1897857 [Crossref] [ Google Scholar]
- Guedan S, Calderon H, Posey Jr, AD AD, Maus M V. Engineering and Design of Chimeric Antigen Receptors. Mol Ther Methods Clin Dev 2019; 12:145-156. doi: 10.1016/j.omtm.2018.12.009 [Crossref] [ Google Scholar]
- Sadelain M, Brentjens R, Rivière I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov 2013; 3:388-398. doi: 10.1158/2159-8290.CD-12-0548 [Crossref] [ Google Scholar]
- Forsberg EM V, Lindberg MF, Jespersen H, Alsén S, Bagge RO, Donia M. HER2 CAR-T Cells Eradicate Uveal Melanoma and T-cell Therapy–Resistant Human Melanoma in IL2 Transgenic NOD/SCID IL2 Receptor Knockout Mice. Cancer Res 2019; 79:899-904. doi: 10.1158/0008-5472.CAN-18-3158 [Crossref] [ Google Scholar]
- Al-Ameer HJ, Zihlif M, Maslat A, Al-Awaida WJ, Ayyash AM, Imraish A. Targeting the proliferation of glioblastoma cells and enhancement of doxorubicin and temozolomide cytotoxicity through inhibition of PFKFB4 and HMOX1 genes with siRNAs. Sci Rep 2025; 15:27861. doi: 10.1038/s41598-025-97192-z [Crossref] [ Google Scholar]
- Grigoruta M, Kong X, Qin Y. Advances and Challenges in Immunotherapy for Metastatic Uveal Melanoma: Clinical Strategies and Emerging Targets. J Clin Med 2025; 14:5137. doi: 10.3390/jcm14145137 [Crossref] [ Google Scholar]
- Leonard-Murali S, Kammula US. Optimizing TIL therapy for uveal melanoma: lessons learned and unlearned from cutaneous melanoma. Immunotherapy 2025; 17:283-291. doi: 10.1080/1750743X.2025.2478808 [Crossref] [ Google Scholar]
- Zugasti I, Espinosa-Aroca Espinosa-Aroca, Lady Lady, Fidyt K, Mulens-Arias V, Diaz-Beya M. CAR-T cell therapy for cancer: current challenges and future directions. Signal Transduct Target Ther 2025; 10:210. doi: 10.1038/s41392-025-02269-w [Crossref] [ Google Scholar]
- Rafii S, Mukherji D, Komaranchath AS, Khalil C, Iqbal F, Abdelwahab SI. Advancing CAR T-Cell Therapy in Solid Tumors: Current Landscape and Future Directions. Cancers (Basel) 2025; 17:2898. doi: 10.3390/cancers17172898 [Crossref] [ Google Scholar]
-
Julve M, Wong YNS, Lim KHJ, Furness AJS. Solid tumour cellular therapy - principles of toxicity management. Immuno-Oncology Technol 2025; 25. doi: 10.1016/j.iotech.2024.100737.
- Parums D V. A Review of CAR T Cells and Adoptive T-Cell Therapies in Lymphoid and Solid Organ Malignancies. Med Sci Monit Int Med J Exp Clin Res 2025; 31:e948125. doi: 10.12659/MSM.948125 [Crossref] [ Google Scholar]
- Haanen J, Los C, Phan GQ, Betof Warner A. Adoptive Cell Therapy for Solid Tumors: Current Status in Melanoma and Next-Generation Therapies. Am Soc Clin Oncol Educ B 2024; 44:e431608. doi: 10.1200/EDBK_431608 [Crossref] [ Google Scholar]
- Kelly E, Russell SJ. History of Oncolytic Viruses: Genesis to Genetic Engineering. Mol Ther 2007; 15:651-659. doi: 10.1038/sj.mt.6300108 [Crossref] [ Google Scholar]
- Southam CM. DIVISION OF MICROBIOLOGY: PRESENT STATUS OF ONCOLYTIC VIRUS STUDIES. Trans N Y Acad Sci 1960; 22:657-673. doi: 10.1111/j.2164-0947.1960.tb00739.x [Crossref] [ Google Scholar]
- Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental Therapy of Human Glioma by Means of a Genetically Engineered Virus Mutant. Science 1991; 252:854-856. doi: 10.1126/science.1851332 [Crossref] [ Google Scholar]
- Au G. G, Lindberg Michael A, Barry D. R, Shafren R. D.. Oncolysis of vascular malignant human melanoma tumors by Coxsackievirus A21. Int J Oncol 2005; 26:1471-1476. doi: 10.3892/ijo.26.6.1471 [Crossref] [ Google Scholar]
- Rudin CM, Poirier JT, Senzer NN, Stephenson Joseph J, Loesch D, Burroughs KD. Phase I Clinical Study of Seneca Valley Virus (SVV-001),a Replication-Competent Picornavirus, in Advanced Solid Tumors with Neuroendocrine Features. Clin Cancer Res 2011; 17:888-895. doi: 10.1158/1078-0432.CCR-10-1706 [Crossref] [ Google Scholar]
- Liu T-C, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol 2007; 4:101-117. doi: 10.1038/ncponc0736 [Crossref] [ Google Scholar]
- Schoofs G, Monica TJ, Ayala J, Horwitz J, Montgomery T, Roth G. A high-yielding serum-free, suspension cell culture process to manufacture recombinant adenoviral vectors for gene therapy. Cytotechnology 1998; 28:81-89. doi: 10.1023/A:1008021428969 [Crossref] [ Google Scholar]
- Knop DR, Harrell H. Bioreactor Production of Recombinant Herpes Simplex Virus Vectors. Biotechnol Prog 2007; 23:715-721. doi: 10.1021/bp060373p [Crossref] [ Google Scholar]
- Lewis JA, Brown EL, Duncan PA. Approaches to the release of a master cell bank of PERC6 cells; a novel cell substrate for the manufacture of human vaccines. Dev Biol (Basel) 2006; 123:165-76. [ Google Scholar]
- Russell SJ. Replicating vectors for cancer therapy: a question of strategy. Semin Cancer Biol 1994; 5:437-443. [ Google Scholar]
- Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G. Phase II Clinical Trial of a Granulocyte-Macrophage Colony-Stimulating Factor–Encoding, Second-Generation Oncolytic Herpesvirus in Patients With Unresectable Metastatic Melanoma. J Clin Oncol 2009; 27:5763-5771. doi: 10.1200/JCO.2009.24.3675 [Crossref] [ Google Scholar]
- Park B-H, Hwang T, Liu T-C, Sze DY, Kim J-S, Kwon H-C. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol 2008; 9:533-542. doi: 10.1016/S1470-2045(08)70107-4 [Crossref] [ Google Scholar]
- Eager RM, Nemunaitis J. Clinical development directions in oncolytic viral therapy. Cancer Gene Ther 2011; 18:305-317. doi: 10.1038/cgt.2011.7 [Crossref] [ Google Scholar]
- Mastrangelo MJ, Maguire HC, Eisenlohr LC, Laughlin CE, Monken CE, McCue PA. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther 1999; 6:409-422. doi: 10.1038/sj.cgt.7700066 [Crossref] [ Google Scholar]
- Harrington KJ, Hingorani M, Tanay MA, Hickey J, Bhide SA, Clarke PM. Phase I/II Study of Oncolytic HSVGM-CSF in Combination with Radiotherapy and Cisplatin in Untreated Stage III/IV Squamous Cell Cancer of the Head and Neck. Clin Cancer Res 2010; 16:4005-4015. doi: 10.1158/1078-0432.CCR-10-0196 [Crossref] [ Google Scholar]
- Harrington KJ, Vile RG, Melcher A, Chester J, Pandha HS. Clinical trials with oncolytic reovirus: Moving beyond phase I into combinations with standard therapeutics. Cytokine Growth Factor Rev 2010; 21:91-98. doi: 10.1016/j.cytogfr.2010.02.006 [Crossref] [ Google Scholar]
- Heo J, Breitbach CJ, Moon A, Kim CW, Patt R, Kim MK. Sequential Therapy With JX-594, A Targeted Oncolytic Poxvirus, Followed by Sorafenib in Hepatocellular Carcinoma: Preclinical and Clinical Demonstration of Combination Efficacy. Mol Ther 2011; 19:1170-1179. doi: 10.1038/mt.2011.39 [Crossref] [ Google Scholar]
- Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQM. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011; 477:99-102. doi: 10.1038/nature10358 [Crossref] [ Google Scholar]
- Serganova I, Ponomarev V, Blasberg R. Human reporter genes: potential use in clinical studies. Nucl Med Biol 2007; 34:791-807. doi: 10.1016/j.nucmedbio.2007.05.009 [Crossref] [ Google Scholar]
- Peng K-W, Facteau S, Wegman T, O’Kane D, Russell SJ. Non-invasive in vivo monitoring of trackable viruses expressing soluble marker peptides. Nat Med 2002; 8:527-531. doi: 10.1038/nm0502-527 [Crossref] [ Google Scholar]
- Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA. Phase I Trial of Intraperitoneal Administration of an Oncolytic Measles Virus Strain Engineered to Express Carcinoembryonic Antigen for Recurrent Ovarian Cancer. Cancer Res 2010; 70:875-882. doi: 10.1158/0008-5472.CAN-09-2762 [Crossref] [ Google Scholar]
- Jacobs A, Voges J, Reszka R, Lercher M, Gossmann A, Kracht L. Positron-emission tomography of vector-mediated gene expression in gene therapy for gliomas. Lancet 2001; 358:727-729. doi: 10.1016/S0140-6736(01)05904-9 [Crossref] [ Google Scholar]
- Barton KN, Stricker H, Brown SL, Elshaikh M, Aref I, Lu M. Phase I Study of Noninvasive Imaging of Adenovirus-mediated Gene Expression in the Human Prostate. Mol Ther 2008; 16:1761-1769. doi: 10.1038/mt.2008.172 [Crossref] [ Google Scholar]
- Dingli D, Peng K-W, Harvey ME, Greipp PR, O’Connor MK, Cattaneo R. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood 2004; 103:1641-1646. doi: 10.1182/blood-2003-07-2233 [Crossref] [ Google Scholar]
- Zarezadeh Mehrabadi A, Tat M, Ghorbani Alvanegh A, Roozbahani F, Esmaeili Gouvarchin Ghaleh H. Revolutionizing cancer treatment: the power of bi- and tri-specific T-cell engagers in oncolytic virotherapy. Front Immunol 2024; 15:1343378. doi: 10.3389/fimmu.2024.1343378 [Crossref] [ Google Scholar]
- Guo ZS, Liu Z, Kowalsky S, Feist M, Kalinski P, Lu B, Storkus WJ. Oncolytic Immunotherapy: Conceptual Evolution, Current Strategies, and Future Perspectives. Front Immunol 2017; 8:555. doi: 10.3389/fimmu.2017.00555 [Crossref] [ Google Scholar]
- Cham LB, Hamdan TA, Bhat H, Sirajo B, Ali M, Tabbara KS. Immunotherapeutic Blockade of CD47 Increases Virus Neutralization Antibodies. Vaccines 2025; 13:602. doi: 10.3390/vaccines13060602 [Crossref] [ Google Scholar]
- Nguyen T, Avci NG, Shin DH, Martinez-Velez N, Jiang H. Tune Up In Situ Autovaccination against Solid Tumors with Oncolytic Viruses. Cancers (Basel) 2018; 10:171. doi: 10.3390/cancers10060171 [Crossref] [ Google Scholar]
- Cook J, Acosta-Medina AA, Peng KW, Lacy M, Russell S. Oncolytic virotherapy - Forging its place in the immunomodulatory paradigm for Multiple Myeloma. Cancer Treat Res Commun 2021; 29:100473. doi: 10.1016/j.ctarc.2021.100473 [Crossref] [ Google Scholar]
- Glorioso JC, Cohen JB, Goins WF, Hall B, Jackson JW, Kohanbash G. Oncolytic HSV Vectors and Anti-Tumor Immunity. Curr Issues Mol Biol 2021; 41:381-468. doi: 10.21775/cimb.041.381 [Crossref] [ Google Scholar]
- Bausch-Fluck D, Hofmann A, Bock T, Frei AP, Cerciello F, Jacobs A. A Mass Spectrometric-Derived Cell Surface Protein Atlas. PLoS One 2015; 10:e0121314. doi: 10.1371/journal.pone.0121314 [Crossref] [ Google Scholar]
- Berman DM, Bell JI. Redirecting Polyclonal T Cells against Cancer with Soluble T-Cell Receptors. Clin Cancer Res 2023; 29:697-704. doi: 10.1158/1078-0432.CCR-22-0028 [Crossref] [ Google Scholar]
- Barrow AD, Martin CJ, Colonna M. The Natural Cytotoxicity Receptors in Health and Disease. Front Immunol 2019; 10:909. doi: 10.3389/fimmu.2019.00909 [Crossref] [ Google Scholar]
- Novotny J, Ganju RK, Smiley ST, Hussey RE, Luther MA, Recny MA. A soluble, single-chain T-cell receptor fragment endowed with antigen-combining properties. Proc Natl Acad Sci 1991; 88:8646-8650. doi: 10.1073/pnas.88.19.8646 [Crossref] [ Google Scholar]
- Dhillon S. Tebentafusp: First Approval. Drugs 2022; 82:703-710. doi: 10.1007/s40265-022-01704-4 [Crossref] [ Google Scholar]
- Augsberger C, Hänel G, Xu W, Pulko V, Hanisch LJ, Augustin A. Targeting intracellular WT1 in AML with a novel RMF-peptide-MHC-specific T-cell bispecific antibody. Blood 2021; 138:2655-2669. doi: 10.1182/blood.2020010477 [Crossref] [ Google Scholar]
- Chervin AS, Stone JD, Konieczna I, Calabrese KM, Wang N, Haribhai D. ABBV-184: A Novel Survivin-specific TCR/CD3 Bispecific T-cell Engager is Active against Both Solid Tumor and Hematologic Malignancies. Mol Cancer Ther 2023; 22:903-912. doi: 10.1158/1535-7163.MCT-22-0770 [Crossref] [ Google Scholar]
- van Diest E, Nicolasen MJT, Kramer L, Zheng J, Hernández-López P, Beringer DX. The making of multivalent gamma delta TCR anti-CD3 bispecific T cell engagers. Front Immunol 2023; 13:1052090. doi: 10.3389/fimmu.2022.1052090 [Crossref] [ Google Scholar]
- Yarmarkovich M, Marshall QF, Warrington JM, Premaratne R, Farrel A, Groff D. RETRACTED ARTICLE: Cross-HLA targeting of intracellular oncoproteins with peptide-centric CARs. Nature 2021; 599:477-484. doi: 10.1038/s41586-021-04061-6 [Crossref] [ Google Scholar]
- Mungalov R V, Mushenkova N V, Chudakov DM, Turchaninova MA. Engaging T cells for cleanup. Front Immunol 2025; 16:1551424. doi: 10.3389/fimmu.2025.1551424 [Crossref] [ Google Scholar]
- Haus-Cohen M, Reiter Y. Harnessing antibody-mediated recognition of the intracellular proteome with T cell receptor-like specificity. Front Immunol 2024; 15:1486721. doi: 10.3389/fimmu.2024.1486721 [Crossref] [ Google Scholar]
- Albayrak G, Wan PK-T, Fisher K, Seymour LW. T cell engagers: expanding horizons in oncology and beyond. Br J Cancer 2025; 133:1241-1249. doi: 10.1038/s41416-025-03125-y [Crossref] [ Google Scholar]
- van der Kooij MK, Kapiteijn E, Rutkowski P, Baurain J-F, de Koning L, Speetjens FM. Real-life data on tebentafusp in metastatic uveal melanoma patients from four EURACAN Expert Centres. Eur J Cancer 2025; 227:115634. doi: 10.1016/j.ejca.2025.115634 [Crossref] [ Google Scholar]
- Wang Y, Sun W, Wang B. Evaluating the efficacy and safety of tebentafusp in the treatment of metastatic uveal melanoma: a 2025 update systematic review and meta-analysis. Front Oncol 2025; 15:1667282. doi: 10.3389/fonc.2025.1667282 [Crossref] [ Google Scholar]
- Zou Y, Kamoi K, Zong Y, Zhang J, Yang M, Ohno-Matsui K. Vaccines and the Eye: Current Understanding of the Molecular and Immunological Effects of Vaccination on the Eye. Int J Mol Sci 2024; 25:4755. doi: 10.3390/ijms25094755 [Crossref] [ Google Scholar]
- Scoles S, Ganesh S, Yamada KH. Current Therapies and Potential Strategies for Uveal Melanoma. Drugs Drug Candidates 2025; 4:14. doi: 10.3390/ddc4020014 [Crossref] [ Google Scholar]
- Masalkhi M, Wahoud N, Moran B, Elhassadi E. Immunotherapy in Ophthalmic Oncology: Current Trends and Future Directions. J Clin TranslOphthalmol 2025; 3:1. doi: 10.3390/jcto3010001 [Crossref] [ Google Scholar]
- Kerley M, Piri N, Ramasubramanian A. Diffuse Pigment Release in a Patient Undergoing Tumor-Infiltrating Lymphocyte Immunotherapy for Acral Malignant Melanoma. J Ophthalmic Vis Res 2023; 18:339-341. doi: 10.18502/jovr.v18i3.13783 [Crossref] [ Google Scholar]
- Murty T, Wai KM, Rahimy E, Mruthyunjaya P. Low Occurrence of Ocular Adverse Events after CAR-T Cell Therapy. Ocul Oncol Pathol 2025; 11:104-108. doi: 10.1159/000543055 [Crossref] [ Google Scholar]
- Kulbay M, Tuli N, Mazza M, Jaffer A, Juntipwong S, Marcotte E. Oncolytic Viruses and Immunotherapy for the Treatment of Uveal Melanoma and Retinoblastoma: The Current Landscape and Novel Advances. Biomedicines 2025; 13:108. doi: 10.3390/biomedicines13010108 [Crossref] [ Google Scholar]
- Wen J, Cui W, Yin X, Chen Y, Liu A, Wang Q. Application and future prospects of bispecific antibodies in the treatment of non-small cell lung cancer. Cancer Biol Med 2025; 22:348-375. doi: 10.20892/j.issn.2095-3941.2024.0470 [Crossref] [ Google Scholar]
- Wang X, Wang T, Lam E, Alvarez D, Sun Y. Ocular Vascular Diseases: From Retinal Immune Privilege to Inflammation. Int J Mol Sci 2023; 24:12090. doi: 10.3390/ijms241512090 [Crossref] [ Google Scholar]
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015; 348:74-80. doi: 10.1126/science.aaa6204 [Crossref] [ Google Scholar]
- Caspi RR. Ocular autoimmunity: the price of privilege?. Immunol Rev 2006; 213:23-35. doi: 10.1111/j.1600-065X.2006.00439.x [Crossref] [ Google Scholar]
- Concepcion C, Xia Y, Korshunova Y, Bligard GW, Taylor A, Paley MA. Compositional variation in eye-infiltrating immune cells distinguishes human uveitis subtypes. iScience 2025; 28:111928. doi: 10.1016/j.isci.2025.111928 [Crossref] [ Google Scholar]
- Hori J, Joyce N, Streilein JW. Epithelium-Deficient Corneal Allografts Display Immune Privilege beneath the Kidney Capsule. Invest Ophthalmol Vis Sci 2000; 41:443-452. [ Google Scholar]
- Hori J, Joyce NC, Streilein JW. Immune Privilege and Immunogenicity Reside among Different Layers of the Mouse Cornea. Invest Ophthalmol Vis Sci 2000; 41:3032-3042. [ Google Scholar]
- Wenkel H, Streilein JW. Evidence that Retinal Pigment Epithelium Functions as an Immune-Privileged Tissue. Invest Ophthalmol Vis Sci 2000; 41:3467-3473. [ Google Scholar]
- Streilein JW. Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol 2003; 3:879-889. doi: 10.1038/nri1224 [Crossref] [ Google Scholar]
- Niederkorn JY, Stein-Streilein J. History and Physiology of Immune Privilege. Ocul Immunol Inflamm 2010; 18:19-23. doi: 10.3109/09273940903564766 [Crossref] [ Google Scholar]
- Niederkorn JY. Corneal Transplantation and Immune Privilege. Int Rev Immunol 2013; 32:57-67. doi: 10.3109/08830185.2012.737877 [Crossref] [ Google Scholar]
- Hori J, Yamaguchi T, Keino H, Hamrah P, Maruyama K. Immune privilege in corneal transplantation. Prog Retin Eye Res 2019; 72:100758. doi: 10.1016/j.preteyeres.2019.04.002 [Crossref] [ Google Scholar]
- Forrester J V, Xu H. Good news–bad news: the Yin and Yang of immune privilege in the eye. Front Immunol 2012; 3:338. doi: 10.3389/fimmu.2012.00338 [Crossref] [ Google Scholar]
- MEDAWAR PB. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol 1948; 29:58-69. [ Google Scholar]
- Chen M, Luo C, Zhao J, Devarajan G, Xu H. Immune regulation in the aging retina. Prog Retin Eye Res 2019; 69:159-172. doi: 10.1016/j.preteyeres.2018.10.003 [Crossref] [ Google Scholar]
- Shakib M, Cunha-Vaz JG. Studies on the permeability of the blood-retinal barrier: IV Junctional complexes of the retinal vessels and their role in the permeability of the blood-retinal barrier. Exp Eye Res 1966; 5:229-IN16. doi: 10.1016/S0014-4835(66)80011-8 [Crossref] [ Google Scholar]
- PEYMAN GA, BOK D. Peroxidase diffusion in the normal and laser-coagulated primate retina. Invest Ophthalmol Vis Sci 1972; 11:35-45. [ Google Scholar]
- Sugita S. Role of ocular pigment epithelial cells in immune privilege. Arch Immunol Ther Exp (Warsz) 2009; 57:263-268. doi: 10.1007/s00005-009-0030-0 [Crossref] [ Google Scholar]
- Mochizuki M, Sugita S, Kamoi K. Immunological homeostasis of the eye. Prog Retin Eye Res 2013; 33:10-27. doi: 10.1016/j.preteyeres.2012.10.002 [Crossref] [ Google Scholar]
- Mendes-Jorge L, Ramos D, Luppo M, Llombart C, Alexandre-Pires G, Nacher V. Scavenger Function of Resident Autofluorescent Perivascular Macrophages and Their Contribution to the Maintenance of the Blood–Retinal Barrier. Invest Ophthalmol Vis Sci 2009; 50:5997-6005. doi: 10.1167/iovs.09-3515 [Crossref] [ Google Scholar]
- Rajesh A, Droho S, Lavine JA. Macrophages in close proximity to the vitreoretinal interface are potential biomarkers of inflammation during retinal vascular disease. J Neuroinflammation 2022; 19:203. doi: 10.1186/s12974-022-02562-3 [Crossref] [ Google Scholar]
- Raviola G. The structural basis of the blood-ocular barriers. Exp Eye Res 1977; 25:27-63. doi: 10.1016/S0014-4835(77)80009-2 [Crossref] [ Google Scholar]
- Cunha-Vaz J. The blood-ocular barriers. SurvOphthalmol 1979; 23:279-296. doi: 10.1016/0039-6257(79)90158-9 [Crossref] [ Google Scholar]
- Coca-Prados M. The Blood-Aqueous Barrier in Health and Disease. J Glaucoma 2014; 23:S36-8. doi: 10.1097/IJG.0000000000000107 [Crossref] [ Google Scholar]
- Cunha-Vaz J. The Blood-Retinal Barrier in the Management of Retinal Disease: EURETINA Award Lecture. Ophthalmologica 2017; 237:1-10. doi: 10.1159/000455809 [Crossref] [ Google Scholar]
- Taylor AW, Ng TF. Negative regulators that mediate ocular immune privilege. J Leukoc Biol 2018; 103:1179-1187. doi: 10.1002/JLB.3MIR0817-337R [Crossref] [ Google Scholar]
- Goel M, Picciani RG, Lee RK, Bhattacharya SK. Aqueous humor dynamics: a review. Open Ophthalmol J 2010; 4:52-59. doi: 10.2174/1874364101004010052 [Crossref] [ Google Scholar]
- Taylor AW. A review of the influence of aqueous humor on immunity. Ocul Immunol Inflamm 2003; 11:231-241. doi: 10.1076/ocii.11.4.231.18269 [Crossref] [ Google Scholar]
- Namba K, Kitaichi N, Nishida T, Taylor AW. Induction of regulatory T cells by the immunomodulating cytokines α-melanocyte-stimulating hormone and transforming growth factor-β2. J Leukoc Biol 2002; 72:946-952. doi: 10.1189/jlb.72.5.946 [Crossref] [ Google Scholar]
- Cousins SW, Trattler WB, Streilein JW. Immune privilege and suppression of immunogenic inflammation in the anterior chamber of the eye. Curr Eye Res 1991; 10:287-297. doi: 10.3109/02713689108996334 [Crossref] [ Google Scholar]
- Nishida T, Taylor AW. Specific aqueous humor factors induce activation of regulatory T cells. Invest Ophthalmol Vis Sci 1999; 40:2268-2274. [ Google Scholar]
- Zamiri P, Masli S, Kitaichi N, Taylor AW, Streilein JW. Thrombospondin plays a vital role in the immune privilege of the eye. Invest Ophthalmol Vis Sci 2005; 46:908-919. doi: 10.1167/iovs.04-0362 [Crossref] [ Google Scholar]
- Lau CH, Taylor AW. The immune privileged retina mediates an alternative activation of J774A 1 cells. Ocul Immunol Inflamm 2009; 17:380-389. doi: 10.3109/09273940903118642 [Crossref] [ Google Scholar]
- Wilbanks GA, Wayne Streilein J. Fluids from immune privileged sites endow macrophages with the capacity to induce antigen‐specific immune deviation via a mechanism involving transforming growth factor‐β. Eur J Immunol 1992; 22:1031-1036. doi: 10.1002/eji.1830220423 [Crossref] [ Google Scholar]
- Vendomèle J, Khebizi Q, Fisson S. Cellular and molecular mechanisms of anterior chamber-associated immune deviation (ACAID): what we have learned from knockout mice. Front Immunol 2017; 8:1686. doi: 10.3389/fimmu.2017.01686 [Crossref] [ Google Scholar]
- Wenkel H, Chen PW, Ksander BR, Streilein JW. Immune privilege is extended, then withdrawn, from allogeneic tumor cell grafts placed in the subretinal space. Invest Ophthalmol Vis Sci 1999; 40:3202-3208. [ Google Scholar]
- Jiang LQ, Jorquera M, Streilein JW. Subretinal space and vitreous cavity as immunologically privileged sites for retinal allografts. Invest Ophthalmol Vis Sci 1993; 34:3347-3354. [ Google Scholar]
- Reyes NJ, O’Koren EG, Saban DR. New insights into mononuclear phagocyte biology from the visual system. Nat Rev Immunol 2017; 17:322-332. doi: 10.1038/nri.2017.13 [Crossref] [ Google Scholar]
- Streilein JW. Ocular immune privilege: the eye takes a dim but practical view of immunity and inflammation. J Leucoc Biol 2003; 74:179-185. doi: 10.1189/jlb.1102574 [Crossref] [ Google Scholar]
- Bairagi RD, Reon RR, Hasan MM, Sarker S, Debnath D, Rahman MT. Ocular drug delivery systems based on nanotechnology: a comprehensive review for the treatment of eye diseases. Discov Nano 2025; 20:75. doi: 10.1186/s11671-025-04234-6 [Crossref] [ Google Scholar]
- Abbasi M, Aghamollaei H, Vaez A, Amani AM, Kamyab H, Chelliapan S. Bringing ophthalmology into the scientific world: Novel nanoparticle-based strategies for ocular drug delivery. Ocul Surf 2025; 37:140-172. doi: 10.1016/j.jtos.2025.03.004 [Crossref] [ Google Scholar]
- Song R, Lin Y, Zhang M, Liu Z, Zhang R, Zhao J. Ocular delivery of lipid nanoparticles-formulated mRNA encoding lanosterol synthase ameliorates cataract in rats. Nat Commun 2025; 16:8522. doi: 10.1038/s41467-025-63553-5 [Crossref] [ Google Scholar]
- Abou-Taleb BA, Abdelwahab IA. Comparative evaluation of nano ocular delivery systems loaded pH and thermosensitive in situ gels for Acanthamoeba keratitis treatment. Sci Rep 2025; 15:19430. doi: 10.1038/s41598-025-03418-5 [Crossref] [ Google Scholar]
- Gade S, So Y, Mishra D, Baviskar SM, Assiri AA, Glover K. Ocular Drug Delivery: Emerging Approaches and Advances. Pharmaceutics 2025; 17:599. doi: 10.3390/pharmaceutics17050599 [Crossref] [ Google Scholar]
- Tsoplaktsoglou M, Spyratou E, Droulias A, Zachou M-E, Efstathopoulos EP. The Contribution of Nanomedicine in Ocular Oncology. Cancers (Basel) 2025; 17:1186. doi: 10.3390/cancers17071186 [Crossref] [ Google Scholar]
- Chhoker D, Yadav AK, Sinha VR. Ocular implants and inserts: revolutionizing drug delivery in ophthalmology. Int J Pharm 2025; 685:126248. doi: 10.1016/j.ijpharm.2025.126248 [Crossref] [ Google Scholar]
- Peng Z, Nagarajan V, Horai R, Jittayasothorn Y, Mattapallil MJ, Caspi RR. Ocular immune privilege in action: The living eye imposes unique regulatory and anergic gene signatures on uveitogenic T cells. Cell Rep 2025; 44:115780. doi: 10.1016/j.celrep.2025.115780 [Crossref] [ Google Scholar]
- Li B, Zhang X, Fang Y, Chen M, Li Q, Zeng Y. PD-L1 Promotes Immunological Tolerance and Enhances Visual Protection of hESC-RPE Grafts in Retinal Degeneration. Cell Prolif 2025; 58:e70007. doi: 10.1111/cpr.70007 [Crossref] [ Google Scholar]
- Ling S, Zhang X, Dai Y, Jiang Z, Zhou X, Lu S. Customizable virus-like particles deliver CRISPR–Cas9 ribonucleoprotein for effective ocular neovascular and Huntington’s disease gene therapy. Nat Nanotechnol 2025; 20:543-553. doi: 10.1038/s41565-024-01851-7 [Crossref] [ Google Scholar]
- Murphy R, Martin KR. Genetic engineering and the eye. Eye 2025; 39:57-68. doi: 10.1038/s41433-024-03441-2 [Crossref] [ Google Scholar]
- Lin X, Zhou Y, Lv K, Wu W, Chen C. Nanomedicine-Based Ophthalmic Drug Delivery Systems for the Treatment of Ocular Diseases. Int J Nanomedicine 2025; 20:9221-9249. doi: 10.2147/IJN.S532074 [Crossref] [ Google Scholar]
- Kapoor DU, Patel G, Prajapati BG. Nanomedicine-Based Treatments for Rare and Aggressive Ocular Cancers: Advances in Drug Delivery. Curr Treat Options Oncol 2025; 26:569-586. doi: 10.1007/s11864-025-01330-8 [Crossref] [ Google Scholar]
- Ibeas Moreno E, Alonso MJ, Abbadessa A. Intraocular injectable hydrogels for the delivery of cells and nanoparticles. Mater Today Bio 2025; 32:101767. doi: 10.1016/j.mtbio.2025.101767 [Crossref] [ Google Scholar]
- Mamidi N, Mahmoudsalehi AO. Advances in three-dimensional hydrogel networks for cancer immunotherapy. J Mater Chem B 2025; 13:10440-10459. doi: 10.1039/D5TB01133G [Crossref] [ Google Scholar]
- Mohammadzadeh V, Atapour-Mashhad H, Shahvali S, Salehi B, Shaban M, Shirzad M. Hydrogels as advanced drug delivery platforms for cancer immunotherapy: promising innovations and future outlook. J Nanobiotechnology 2025; 23:545. doi: 10.1186/s12951-025-03613-6 [Crossref] [ Google Scholar]
- Su Y, Chen M, Xu W, Gu P, Fan X. Advances in Extracellular-Vesicles-Based Diagnostic and Therapeutic Approaches for Ocular Diseases. ACS Nano 2024; 18:22793-22828. doi: 10.1021/acsnano.4c08486 [Crossref] [ Google Scholar]
- Ou S, Lin Y, Zhang Y, Shi K, Wu H. Epidemiology and tumor microenvironment of ocular surface and orbital tumors on growth and malignant transformation. Front Oncol 2024; 14:1388156. doi: 10.3389/fonc.2024.1388156 [Crossref] [ Google Scholar]
- Abdelmohsen HAM, Copeland NA, Hardy JG. Light-responsive biomaterials for ocular drug delivery. Drug DelivTransl Res 2023; 13:2159-2182. doi: 10.1007/s13346-022-01196-5 [Crossref] [ Google Scholar]
- Fu W, Lei C, Wang C, Ma Z, Li T, Lin F. Synthetic libraries of immune cells displaying a diverse repertoire of chimaeric antigen receptors as a potent cancer immunotherapy. Nat Biomed Eng 2022; 6:842-854. doi: 10.1038/s41551-022-00895-1 [Crossref] [ Google Scholar]
-
Ansari M, Shinde U, Singh K. 19 - Ocular cancer—Current and new drug delivery systems. In: Shegokar R, Pathak YV, eds. Drug Delivery Landscape in Cancer Research. Academic Press; 2025. p. 451–472. doi: 10.1016/B978-0-443-29168-5.00020-9.
- Wu KY, Wang XC, Anderson M, Tran SD. Advancements in Nanosystems for Ocular Drug Delivery: A Focus on Pediatric Retinoblastoma. Molecules 2024; 29:2263. doi: 10.3390/molecules29102263 [Crossref] [ Google Scholar]
- Jang G-F, Crabb JS, Hu B, Willard B, Kalirai H, Singh AD. Proteomics of primary uveal melanoma: insights into metastasis and protein biomarkers. Cancers (Basel) 2021; 13:3520. doi: 10.3390/cancers13143520 [Crossref] [ Google Scholar]
-
Song J, Zhang Z, Chan DW. Detection of Uveal Melanoma by Multiplex Immunoassays of Serum Biomarkers. In: Melanoma: Methods and Protocols. Springer; 2021. p. 447–459. doi: 10.1007/978-1-0716-1205-7_32.
- Song J, Merbs SL, Sokoll LJ, Chan DW, Zhang Z. A multiplex immunoassay of serum biomarkers for the detection of uveal melanoma. Clin Proteomics 2019; 16:10. doi: 10.1186/s12014-019-9230-8 [Crossref] [ Google Scholar]
- Velez G, Nguyen H V, Chemudupati T, Ludwig CA, Toral M, Reddy S. Liquid biopsy proteomics of uveal melanoma reveals biomarkers associated with metastatic risk. Mol Cancer 2021; 20:39. doi: 10.1186/s12943-021-01336-4 [Crossref] [ Google Scholar]
- Rodrigues M, Ramtohul T, Rampanou A, Sandoval JL, Houy A, Servois V. Prospective assessment of circulating tumor DNA in patients with metastatic uveal melanoma treated with tebentafusp. Nat Commun 2024; 15:8851. doi: 10.1038/s41467-024-53145-0 [Crossref] [ Google Scholar]
- Sanchez GM, Chigane D, Lin M, Xu L, Yellapantula V, Berry JL. Retinoblastoma: Aqueous humor liquid biopsy. Taiwan J Ophthalmol 2025; 15:55-61. doi: 10.4103/tjo.TJO-D-24-00133 [Crossref] [ Google Scholar]
- Li H-T, Xu L, Weisenberger DJ, Li M, Zhou W, Peng C-C. Characterizing DNA methylation signatures of retinoblastoma using aqueous humor liquid biopsy. Nat Commun 2022; 13:5523. doi: 10.1038/s41467-022-33248-2 [Crossref] [ Google Scholar]
- Shen X, Zhou H, Zhou X, Liu Z, Meng X, Zhang L. 68Ga-grazytracer PET for noninvasive assessment of response to immunotherapy in solid tumors and lymphomas: a phase 1/2 clinical trial. Nat Commun 2024; 15:8791. doi: 10.1038/s41467-024-53197-2 [Crossref] [ Google Scholar]
- Boettcher AN. Preclinical Evaluation of a Granzyme B PET Tracer for the Assessment of Immunotherapy Response. Radiol Imaging cancer 2021; 3:e219001. doi: 10.1148/rycan.2021219001 [Crossref] [ Google Scholar]
- Martens A, Schauwvlieghe PP, Madoe A, Casteels I, Aspeslagh S. Ocular adverse events associated with immune checkpoint inhibitors, a scoping review. J Ophthalmic Inflamm Infect 2023; 13:5. doi: 10.1186/s12348-022-00321-2 [Crossref] [ Google Scholar]
- LaSalle T, Austin EE, Rigney G, Wehrenberg-Klee E, Nesti S, Larimer B. Granzyme B PET imaging of immune-mediated tumor killing as a tool for understanding immunotherapy response. J Immunother Cancer 2020; 8:e000291. doi: 10.1136/jitc-2019-000291 [Crossref] [ Google Scholar]
- Hartimath S V, Ramasamy B, Xuan TY, Rong TJ, Khanapur S, Cheng P. Granzyme B PET Imaging in Response to In Situ Vaccine Therapy Combined with αPD1 in a Murine Colon Cancer Model. Pharmaceutics 2022; 14:150. doi: 10.3390/pharmaceutics14010150 [Crossref] [ Google Scholar]
- Naseripour M, Ghasemi Falavarjani K, Mirshahi R, Sedaghat A. Optical coherence tomography angiography (OCTA) applications in ocular oncology. Eye (Lond) 2020; 34:1535-1545. doi: 10.1038/s41433-020-0819-y [Crossref] [ Google Scholar]
- Obuchowska I, Konopińska J. Importance of Optical Coherence Tomography and Optical Coherence Tomography Angiography in the Imaging and Differentiation of Choroidal Melanoma: A Review. Cancers (Basel) 2022; 14:3354. doi: 10.3390/cancers14143354 [Crossref] [ Google Scholar]
- Yang L, Chen Y, Ling S, Wang J, Wang G, Zhang B. Research progress on the application of optical coherence tomography in the field of oncology. Front Oncol 2022; 12:953934. doi: 10.3389/fonc.2022.953934 [Crossref] [ Google Scholar]
- Zameer MZ, Jou E, Middleton M. The role of circulating tumor DNA in melanomas of the uveal tract. Front Immunol 2024; 15:1509968. doi: 10.3389/fimmu.2024.1509968 [Crossref] [ Google Scholar]
- Barwinski N, Lever M, Rating P, Jabbarli L, Fiorentzis M, Bechrakis NE. Presence of tumor DNA in aqueous humor is correlated with high risk uveal melanoma. Sci Rep 2025; 15:1 9406. doi: 10.1038/s41598-025-03915-7 [Crossref] [ Google Scholar]
- Daneshpour Moghadam S, Maris B, Mokhtari A, Daffara C, Fiorini P. OCT in Oncology and Precision Medicine: From Nanoparticles to Advanced Technologies and AI. Bioengineering (Basel) 2025; 12:650. doi: 10.3390/bioengineering12060650 [Crossref] [ Google Scholar]
- Herskowitz WR, De Arrigunaga S, Greenfield JA, Cohen NK, Galor A, Karp CL. Can high-resolution optical coherence tomography provide an optical biopsy for ocular surface lesions?. Can J Ophthalmol 2025; 60:e185-e196. doi: 10.1016/j.jcjo.2024.07.001 [Crossref] [ Google Scholar]
-
Burnier J V, Abdouh M, Mastromonaco C, Lasiste JM, Burnier MN. Animal Models in Uveal Melanoma. In: Damato BE, Singh AD, eds. Clinical Ophthalmic Oncology: Uveal Tumors. Cham: Springer Nature Switzerland; 2025. p. 125–141. doi: 10.1007/978-3-031-81354-2_11.
- Mukit FA, Terfloth NE, Kim IK, Wu F, Stagner AM. Malignant Transformation of Ocular Melanocytoma to Uveal Melanoma: A Clinicopathologic Review of Three Novel Unsuspected Cases of a Likely Under-Recognized Phenomenon, With Review of the Literature and Molecular Genetic Data. Am J Ophthalmol 2025; 276:126-145. doi: 10.1016/j.ajo.2025.03.044 [Crossref] [ Google Scholar]
- Aram C, Firuzpour F, Barancheshmeh M, Kamali MJ. Unveiling the translational and therapeutic potential of small interfering RNA molecules in combating SARS-CoV-2: A review. Int J Biol Macromol 2025; 318:145203. doi: 10.1016/j.ijbiomac.2025.145203 [Crossref] [ Google Scholar]
- Nazarova V V, Orlova K V, Magomedova ZR, Romanov DS, Maskalenka R, Yarovoy AA. Combined stereotactic radiation therapy and immunotherapy for metastatic uveal melanoma. Front Oncol 2025; 15:1567504. doi: 10.3389/fonc.2025.1567504 [Crossref] [ Google Scholar]
-
Jonas RA, Rokohl AC, Heindl LM. Targeted therapy for malignant ocular melanomas. Ann Eye Sci 2020; 6. doi: 10.21037/aes-20-101.
- Tariq F, Hehar NK, Chigbu DI. The Ocular Surface Microbiome in Homeostasis and Dysbiosis. Microorganisms 2025; 13:1992. doi: 10.3390/microorganisms13091992 [Crossref] [ Google Scholar]
- Kumar NR, Khamar P, Kannan R, Padmanabhan A, Shetty R, D’Souza S. Distinct Ocular Surface Microbiome in Keratoconus Patients Correlate With Local Immune Dysregulation. Invest Ophthalmol Vis Sci 2025; 66:60. doi: 10.1167/iovs.66.1.60 [Crossref] [ Google Scholar]
- Dayhimi Amir, Jazi Kimia, Bigdeli Asiye, Farhoudi Fatemeh, Erfanimanesh Mohamadreza, Esmaeili Mina. Harnessing the Gut Microbiome in Cancer Immunotherapy: Mechanisms, Challenges, and Routes to Personalized Medicine—A Systematic Review. Technol Cancer Res Treat 2025; 24:15330338251365700. doi: 10.1177/15330338251365700 [Crossref] [ Google Scholar]
- Son YM, Kim J. The microbiome-immune axis therapeutic effects in cancer treatments. J Microbiol Biotechnol 2022; 32:1086. doi: 10.4014/jmb.2208.08002 [Crossref] [ Google Scholar]
- Wang R, Li W, Cao H, Zhang L. Decoding the Tumor-Associated Microbiota: From Origins to Nanomedicine Applications in Cancer Therapy. Biology (Basel) 2025; 14:243. doi: 10.3390/biology14030243 [Crossref] [ Google Scholar]
- Borji A, Aram C, Ziyadloo F, Zadeh MR, Rouzbahani KA, Kazemi M. Gene regulation by non-Coding RNAs in infertility: a mechanistic review. J Ovarian Res 2025; 18:265. doi: 10.1186/s13048-025-01862-5 [Crossref] [ Google Scholar]
- Olawade DB, Clement David-Olawade A, Adereni T, Egbon E, Teke J, Boussios S. Integrating AI into Cancer Immunotherapy—A Narrative Review of Current Applications and Future Directions. Diseases 2025; 13:24. doi: 10.3390/diseases13010024 [Crossref] [ Google Scholar]
- Aram C, Karami L, Ranjbar MM. Development of a candidate mRNA vaccine based on Multi-Peptide targeting VP4 of rotavirus A: an immunoinformatics and molecular dynamics approach. Sci Rep 2025; 15:22610. doi: 10.1038/s41598-025-07433-4 [Crossref] [ Google Scholar]
- Firuzpour F, Heydari M, Aram C, Alishvandi A. The role of artificial intelligence in enhancing breast cancer screening and diagnosis: A review of current advances. Bioimpacts 2025; 15:30984. doi: 10.34172/bi.30984 [Crossref] [ Google Scholar]
- Lalman C, Yang Y, Walker JL. Artificial Intelligence in Ocular Transcriptomics: Applications of Unsupervised and Supervised Learning. Cells 2025; 14:1315. doi: 10.3390/cells14171315 [Crossref] [ Google Scholar]
- Fountzilas E, Pearce T, Baysal MA, Chakraborty A, Tsimberidou AM. Convergence of evolving artificial intelligence and machine learning techniques in precision oncology. NPJ Digit Med 2025; 8:75. doi: 10.1038/s41746-025-01471-y [Crossref] [ Google Scholar]
- Karamanli K-E, Maliagkani E, Petrou P, Papageorgiou E, Georgalas I. Artificial Intelligence in Decoding Ocular Enigmas: A Literature Review of Choroidal Nevus and Choroidal Melanoma Assessment. Appl Sci 2025; 15:3565. doi: 10.3390/app15073565 [Crossref] [ Google Scholar]
- Liu J, Fu R, Su Y, Li Z, Huang X, Wang Q. Applications of artificial intelligence in cancer immunotherapy: a frontier review on enhancing treatment efficacy and safety. Front Immunol 2025; 16:1676112. doi: 10.3389/fimmu.2025.1676112 [Crossref] [ Google Scholar]
- Sutanto H, Fetarayani D. Integrating artificial intelligence into small molecule development for precision cancer immunomodulation therapy. NPJ Drug Discov 2025; 2:25. doi: 10.1038/s44386-025-00029-y [Crossref] [ Google Scholar]
-
Pelayes DE, Singh AD. Machine Learning Studies in Ocular Oncology. In: Grzybowski A, ed. Artificial Intelligence in Ophthalmology. Cham: Springer Nature Switzerland; 2025. p. 461–468. doi: 10.1007/978-3-031-83756-2_30.
- Elemam NM, Zhang P, Wu W, Wang Q, Song J, Youness RA. Editorial: Advancements in multi-omics and bioinformatics for the management of solid malignancies. Front Immunol 2025; 16:1640571. doi: 10.3389/fimmu.2025.1640571 [Crossref] [ Google Scholar]
- Jendrichovsky P, Khosravi S, Rupasinghe A, Maximov K, Guo P, Babadi B. Patchy harmonic functional connectivity of the mouse auditory cortex. Proc Natl Acad Sci 2025; 122:e2510012122. doi: 10.1073/pnas.2510012122 [Crossref] [ Google Scholar]
- Zhu Y, Anania VG, Lill JR, Modrusan Z. Editorial: Revolutionizing immunological disease understanding through single cell multi-omics technologies. Front Immunol 2025; 16:1628120. doi: 10.3389/fimmu.2025.1628120 [Crossref] [ Google Scholar]
- Baena JC, Victoria JS, Toro-Pedroza A, Aragón CC, Ortiz-Guzman J, Garcia-Robledo JE. Smart CAR-T Nanosymbionts: archetypes and proto-models. Front Immunol 2025; 16:1635159. doi: 10.3389/fimmu.2025.1635159 [Crossref] [ Google Scholar]
- Imani S, Li X, Chen K, Maghsoudloo M, Jabbarzadeh Kaboli P, Hashemi M. Computational biology and artificial intelligence in mRNA vaccine design for cancer immunotherapy. Front Cell Infect Microbiol 2025; 14:1501010. doi: 10.3389/fcimb.2024.1501010 [Crossref] [ Google Scholar]
- Barua S, Paller CJ, Randhawa N, Rao A. Editorial: AI in digital oncology: imaging and wearable technology for cancer detection and management. Front ArtifIntell 2025; 8:1662971. doi: 10.3389/frai.2025.1662971 [Crossref] [ Google Scholar]
-
Khosravi S, Rupasinghe A, Babadi B. Granger causal inference from spiking observations via latent variable modeling. 2022 56th Asilomar Conference on Signals, Systems, and Computers. IEEE; 2022. p. 618–622. doi: 10.1109/IEEECONF56349.2022.10051886.
- Khanal P, Chikhale R, Machhi J. Editorial: Targeting neuroinflammation for novel therapeutics in neurodegenerative diseases. Front Pharmacol 2025; 16:1602495. doi: 10.3389/fphar.2025.1602495 [Crossref] [ Google Scholar]
- Varshney M, Gehlot A, Sharma A. The synergy of artificial intelligence in biomaterials, regenerative medicine and drug delivery. Next Bioeng 2025; 1:100001. doi: 10.1016/j.nxbio.2025.100001 [Crossref] [ Google Scholar]
- Chen S, Bai W. Artificial intelligence technology in ophthalmology public health: current applications and future directions. Front Cell Dev Biol 2025; 13:1576465. doi: 10.3389/fcell.2025.1576465 [Crossref] [ Google Scholar]
- Bhushan A, Misra P. Unlocking the potential: multimodal AI in biotechnology and digital medicine—economic impact and ethical challenges. npj Digit Med 2025; 8:619. doi: 10.1038/s41746-025-01992-6 [Crossref] [ Google Scholar]
- Tettey-Engmann F, Parupelli SK, Bauer SR, Bhattarai N, Desai S. Advances in Artificial Intelligence-Based Medical Devices for Healthcare Applications. Biomed Mater Devices 2025; 4:1767-87. doi: 10.1007/s44174-025-00379-1 [Crossref] [ Google Scholar]
- Kazemzadeh K. Artificial intelligence in ophthalmology: opportunities, challenges, and ethical considerations. Med hypothesis, DiscovInnovOphthalmol J 2025; 14:255-272. doi: 10.51329/mehdiophthal1517 [Crossref] [ Google Scholar]
- Zhang W, Zeng X, Deng X, Yang F, Ma X, Gao W. Smart biomaterials: as active immune modulators to shape pro-regenerative microenvironments. Front Cell Dev Biol 2025; 13:1669399. doi: 10.3389/fcell.2025.1669399 [Crossref] [ Google Scholar]
- Sharma PK, Chen C-Y. AI-Integrated Micro/Nanorobots for Biomedical Applications: Recent Advances in Design, Fabrication, and Functions. Biosensors 2025; 15:793. doi: 10.3390/bios15120793 [Crossref] [ Google Scholar]
- Vega Escobar K, Armijos PO, Milman T, Shields CL, Eagle RCJ. Intratumoral bacteria in uveal melanoma: A case report. Am J Ophthalmol Case Rep 2023; 30:101833. doi: 10.1016/j.ajoc.2023.101833 [Crossref] [ Google Scholar]
- Fortman DD, Hurd D, Davar D. The Microbiome in Advanced Melanoma: Where Are We Now?. Curr Oncol Rep 2023; 25:997-1016. doi: 10.1007/s11912-023-01431-3 [Crossref] [ Google Scholar]
- Wen Q, Qiu L, Qiu C, Che K, Zeng R, Wang X. Artificial intelligence in predicting efficacy and toxicity of Immunotherapy: Applications, challenges, and future directions. Cancer Lett 2025; 630:217881. doi: 10.1016/j.canlet.2025.217881 [Crossref] [ Google Scholar]
- Koch EAT, Heppt M V, Berking C. The Current State of Systemic Therapy of Metastatic Uveal Melanoma. Am J Clin Dermatol 2024; 25:691-700. doi: 10.1007/s40257-024-00872-1 [Crossref] [ Google Scholar]
- Dimitriou F, Orloff MM, Koch Hein EC, Cheng PF, Hughes IF, Simeone E. Treatment sequence with tebentafusp and immune checkpoint inhibitors in patients with metastatic uveal melanoma and metastatic GNA11/GNAQ mutant melanocytic tumors. Eur J Cancer 2025; 214:115161. doi: 10.1016/j.ejca.2024.115161 [Crossref] [ Google Scholar]
- Park J, Skålhegg BS. Combination of PD-1/PD-L1 and CTLA-4 inhibitors in the treatment of cancer - a brief update. Front Immunol 2025; 16:1680838. doi: 10.3389/fimmu.2025.1680838 [Crossref] [ Google Scholar]
- Jani HS, Ranch K, Pandya R, Patel Y, Boddu SHS, Tiwari AK. An Update on Novel Drug Delivery Systems for the Management of Glaucoma. Pharmaceutics 2025; 17:1087. doi: 10.3390/pharmaceutics17081087 [Crossref] [ Google Scholar]