Bioimpacts. 15:31475.
doi: 10.34172/bi.31475
Review
Allosteric ligand-driven smart nanoconjugates for mutation-selective EGFR targeting: A precision approach to overcoming tyrosine kinase inhibitor resistance
Dilpreet Singh Conceptualization, Investigation, Methodology, Project administration, Writing – original draft, 1, * 
Akshay Kumar Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing, 1
Vir Vikram Sharma Resources, Supervision, Validation, Writing – review & editing, 1
Author information:
1School of Pharmaceutical Sciences, CT University, Ferozepur Rd, Sidhwan Khurd, Punjab 142024, India
Abstract
The development of targeted therapies against epidermal growth factor receptor (EGFR) has transformed the clinical management of EGFR-driven malignancies, especially non-small cell lung cancer (NSCLC). However, the therapeutic benefit of ATP-competitive tyrosine kinase inhibitors (TKIs) is often undermined by acquired resistance mutations such as T790M and C797S, which either enhance ATP affinity or preclude covalent drug binding. Allosteric inhibition of EGFR has emerged as a promising alternative, leveraging cryptic, mutation-specific binding pockets to achieve superior selectivity and reduced off-target toxicity. Allosteric ligands, particularly those targeting the αC-helix adjacent clefts, have shown potent activity against drug-resistant EGFR isoforms but suffer from suboptimal pharmacokinetics and systemic stability. To overcome these limitations, smart nanoconjugates functionalized with allosteric inhibitors have been developed to enhance targeted delivery, improve intracellular trafficking, and facilitate stimuli-responsive drug release. These nanosystems are capable of co-delivering synergistic agents such as siRNA or CRISPR-Cas9 payloads, amplifying pathway suppression and delaying resistance onset. Surface modification strategies, including PEGylation and bioorthogonal ligand conjugation, further improve circulation half-life and tumor accumulation via active and passive targeting. This review systematically discusses the molecular basis of EGFR allosteric inhibition, engineering principles of nanocarrier platforms, including immunogenicity, scale-up feasibility, and regulatory complexities.
Keywords: EGFR mutations, Allosteric inhibitors, Smart nanoconjugates, Targeted drug delivery, Tyrosine kinase resistance, siRNA co-delivery
Copyright and License Information
© 2025 The Author(s).
This work is published by BioImpacts as an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (
http://creativecommons.org/licenses/by-nc/4.0/). Non-commercial uses of the work are permitted, provided the original work is properly cited.
Funding Statement
No specific funding was received for this study.
Introduction
Malignant neoplasms, or cancers, remain a leading cause of morbidity and mortality worldwide, with lung, breast, colorectal, and liver cancers accounting for a significant proportion of the global cancer burden. Despite substantial advancements in chemotherapy, targeted therapy, and immunotherapy, many solid tumors exhibit therapeutic resistance, dose-limiting toxicities, and molecular heterogeneity that hinder long-term disease control. Particularly in oncogene-driven malignancies such as non-small cell lung cancer (NSCLC), therapeutic responses are often transient due to acquired mutations, bypass signaling, or clonal evolution.1,2 The development of molecularly precise therapies that can selectively target aberrant signaling nodes without affecting normal tissues is therefore a critical unmet need in oncology. Against this backdrop, the epidermal growth factor receptor (EGFR) has emerged as a clinically validated but therapeutically challenging target, owing to its dynamic mutational landscape and tendency to develop resistance to ATP-competitive inhibitors.3 This review highlights the emerging strategy of using allosteric ligand-functionalized smart nanoconjugates for mutation-selective EGFR targeting, offering an integrated solution to overcome resistance, enhance tumor selectivity, and enable combinatorial therapeutic delivery. Over the past two decades, EGFR has emerged as a prototypical molecular target, and the advent of ATP-competitive tyrosine kinase inhibitors (TKIs)—such as gefitinib, erlotinib, afatinib, and osimertinib—has substantially improved patient outcomes.4 Nevertheless, clinical efficacy is frequently transient due to the rapid development of resistance mutations such as T790M, C797S, and L718Q, which confer either steric hindrance, increased ATP affinity, or loss of covalent binding potential.5 Allosteric inhibitors target non-canonical binding sites that emerge due to conformational changes in mutant EGFR isoforms, enabling isoform-specific inhibition while sparing wild-type receptors while sparing wild-type receptors—thus minimizing dose-limiting toxicities and enhancing therapeutic precision.6 Recent advances in structural biology and molecular dynamics have enabled rational design of allosteric inhibitors with high binding affinity and configurational adaptability. However, the intrinsic instability, poor bioavailability, and systemic clearance of small-molecule allosteric agents necessitate the development of optimized delivery systems.6,7
To address these challenges, the field has increasingly converged on smart nanoconjugates—nanoscale drug delivery platforms that integrate chemical targeting ligands, responsive release mechanisms, and multivalent architectures to enhance tumor specificity and intracellular delivery.8 Among these, allosteric ligand-functionalized nanocarriers represent a particularly promising innovation, capable of both passive tumor accumulation via the EPR effect and active engagement of mutant EGFR receptors through high-affinity interactions.8,9 First, these nanocarriers vehicles protect fragile allosteric ligands from enzymatic degradation and rapid renal clearance, thereby extending systemic half-life. Second, nanocarriers enable preferential accumulation in tumor tissues through the enhanced EPR effect, while functional surface ligands can facilitate active recognition of mutant EGFR isoforms. Hence, the current investigation is situated at this critical interface of molecular pharmacology and nanotechnology.10 It aims to synthesize recent developments in allosteric EGFR inhibition and highlight how functionalized nanoconjugates offer a precision strategy to overcome drug resistance.11,12,13 This review consolidates the mechanistic rationale, design principles, and translational challenges of allosteric ligand-driven nanomedicine and proposes a framework for next-generation EGFR-targeted therapies in resistant NSCLC.
EGFR mutational landscape and therapeutic resistance
The mutational landscape of EGFR is central to the pathogenesis, progression, and therapeutic responsiveness of multiple cancers, particularly NSCLC, where EGFR mutations are prevalent in up to 50% of East Asian and 15% of Western patients with adenocarcinoma.12 The mutational landscape of EGFR and the structural basis of resistance are summarized in Fig. 1, which depicts the spatial positioning of key mutations (L858R, T790M, C797S) and their effects on ATP binding and conformational dynamics. The most common activating mutations—exon 19 deletions and the L858R point mutation in exon 21—lead to constitutive kinase activation, making EGFR an ideal target for first-generation reversible TKIs such as gefitinib and erlotinib.12,13 However, the clinical efficacy of these agents is transient due to the inevitable emergence of secondary resistance mutations, the most notorious being T790M in exon 20, which increases ATP affinity and sterically hinders inhibitor binding.12 Third-generation TKIs like osimertinib were developed to selectively inhibit T790M mutants while sparing wild-type EGFR, yet resistance even to these agents has been documented, often via C797S mutation that abrogates covalent binding.14,15 Additionally, bypass signaling through MET amplification, HER2 overexpression, PIK3CA activation, and phenotypic transformation into small cell lung cancer or epithelial–mesenchymal transition (EMT) contribute to therapeutic escape.15 These complexities underscore the necessity for mutation-selective therapeutic approaches that can adapt to the heterogeneity and plasticity of EGFR-driven cancers.16 Recent studies have highlighted the potential of allosteric inhibitors and bi-specific molecules that target mutant-specific conformational states without affecting wild-type EGFR, offering a safer and more durable strategy. Moreover, the integration of next-generation sequencing (NGS) for real-time mutation profiling has facilitated the development of dynamic treatment algorithms and molecularly guided drug delivery systems.17 These insights emphasize the importance of designing smart nanoconjugates that are functionally responsive to specific EGFR mutation signatures and capable of circumventing multi-level resistance mechanisms, laying the groundwork for more personalized and adaptive targeted therapies. The clinical relevance of specific EGFR mutations—such as L858R, T790M, and C797S—lies in their structural impact on kinase function and their differential sensitivity to various generations of TKIs (Table 1).
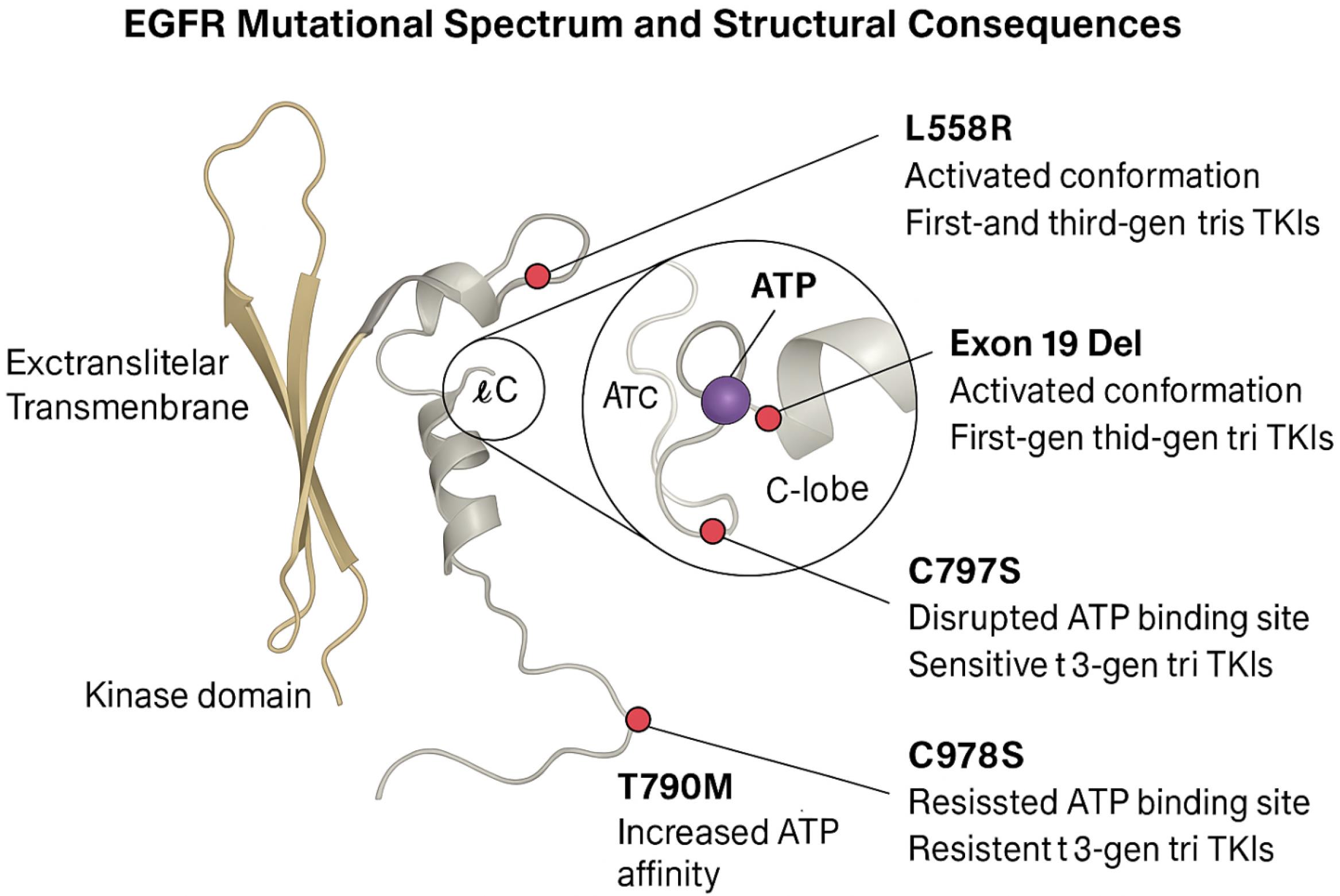
Fig. 1.
Structural mapping of key EGFR mutations (L858R, T790M, C797S) and their influence on kinase activity, ATP binding, and resistance mechanisms.
.
Structural mapping of key EGFR mutations (L858R, T790M, C797S) and their influence on kinase activity, ATP binding, and resistance mechanisms.
Table 1.
Classification of EGFR mutations and associated drug resistance mechanisms
|
Mutation
|
Exon
|
Type
|
Functional impact
|
1st Gen TKIs
|
3rd Gen TKIs
|
Clinical frequency (%)
|
| L858R |
21 |
Activating |
↑ Kinase activity |
Sensitive |
Sensitive |
~30 |
| Exon 19 Del |
19 |
Activating |
Structural activation of TK domain |
Sensitive |
Sensitive |
~45 |
| T790M |
20 |
Resistance |
↑ ATP affinity; steric hindrance |
Resistant |
Sensitive |
~50 (in resistant cases) |
| C797S |
20 |
Resistance |
Blocks covalent binding of 3rd-gen TKIs |
Sensitive |
Resistant |
~20 (post-osimertinib) |
| G719A/C/S |
18 |
Rare Activating |
Partial constitutive activation |
Moderate |
Variable |
~3 |
| S768I |
20 |
Rare Activating |
Alters activation loop |
Moderate |
Variable |
~1 |
| L861Q |
21 |
Rare Activating |
Conformational change in ATP-binding site |
Sensitive |
Sensitive |
~2 |
Allosteric modulation of EGFR: Mechanistic insights
The concept of allosteric modulation in the context of EGFR (epidermal growth factor receptor) therapy has gained significant traction as a means to overcome resistance mechanisms associated with ATP-competitive TKIs. Allosteric inhibitors bind to regions of the EGFR kinase domain that are topographically distinct from the ATP-binding orthosteric pocket, inducing conformational alterations that impede kinase activity without directly competing for ATP.18 A mechanistic comparison of allosteric inhibitors versus ATP-competitive TKIs is illustrated in Fig. 2, highlighting how allosteric agents bind outside the catalytic pocket to stabilize the inactive conformation in mutant receptors. Structural studies using X-ray crystallography and cryo-EM have elucidated key allosteric sites, notably near the αC-helix and activation loop (A-loop), which serve as anchor points for small molecules capable of stabilizing inactive kinase conformations.19 The landmark discovery of EAI045, a mutant-selective allosteric EGFR inhibitor, demonstrated selective efficacy against the T790M and L858R mutations while sparing wild-type EGFR, suggesting a paradigm shift toward safer and mutation-specific therapeutics.20 Several allosteric EGFR inhibitors, including EAI045 and JBJ-04-125-02, have demonstrated nanomolar potency against drug-resistant EGFR isoforms while exhibiting minimal activity against wild-type EGFR (Table 2). Their structural diversity and non-ATP competitive binding profiles make them ideal candidates for conjugation to nanosystems.21 These inhibitors often exhibit synergism with ATP-site binders due to non-overlapping binding modalities, effectively locking the kinase in a catalytically inactive state. Moreover, allosteric binding tends to be less affected by mutations that elevate ATP affinity, such as T790M, making this approach particularly suitable for drug-resistant NSCLC.22 Recent molecular dynamics (MD) simulations and free energy perturbation studies further support the notion that allosteric inhibitors exert their action by disrupting the hydrophobic spine and DFG-in motif alignment, critical for kinase activation.22 The exploitation of these structurally plastic regions has opened new avenues for next-generation inhibitors, and their integration into smart delivery systems—especially ligand-decorated nanocarriers—offers the potential to enhance selectivity, bioavailability, and therapeutic index.23 Collectively, allosteric modulation represents a refined molecular strategy that aligns with the principles of precision oncology and sets the foundation for advanced nanoconjugate-based EGFR-targeted interventions.
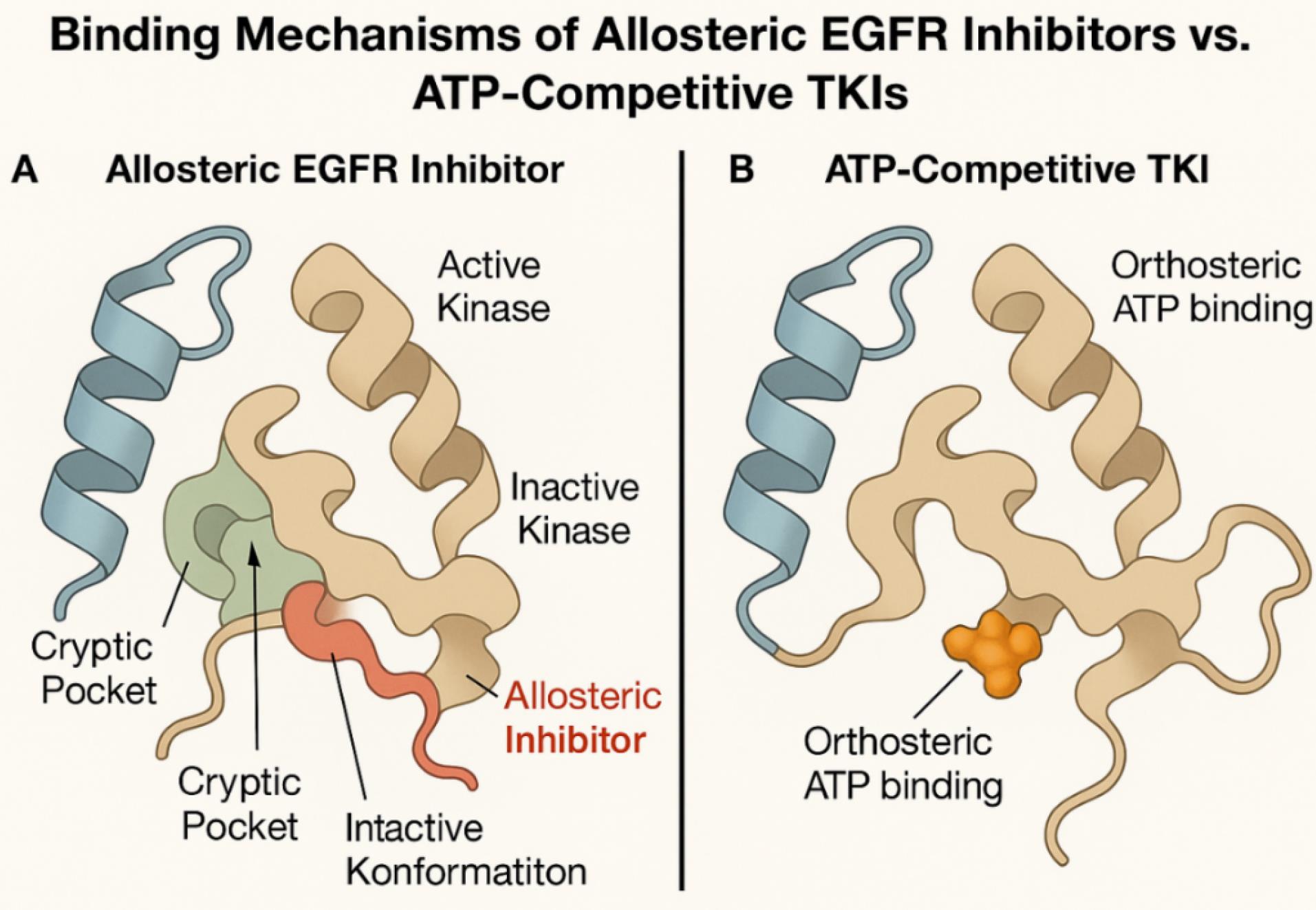
Fig. 2.
Mechanistic illustration comparing the binding of ATP-competitive TKIs (orthosteric inhibitors) versus allosteric EGFR inhibitors. Allosteric inhibitors engage conformationally distinct sites adjacent to the αC-helix, stabilizing the inactive form of mutant EGFR without competing for ATP.
.
Mechanistic illustration comparing the binding of ATP-competitive TKIs (orthosteric inhibitors) versus allosteric EGFR inhibitors. Allosteric inhibitors engage conformationally distinct sites adjacent to the αC-helix, stabilizing the inactive form of mutant EGFR without competing for ATP.
Table 2.
Structural features of reported allosteric EGFR inhibitors
|
Inhibitor
|
Targeted Mutations
|
Allosteric Binding Site
|
Binding Mode
|
Half-maximal inhibitory
concentration (IC₅₀)
(Mutant EGFR)
|
WT EGFR Activity
|
Clinical Stage
|
| EAI045 |
T790M, L858R |
Adjacent to αC-helix (allosteric pocket) |
Non-competitive |
~40 nM |
Inactive |
Preclinical |
| JBJ-04-125-02 |
T790M, C797S |
Allosteric cleft near ATP pocket |
Allosteric-only |
~20 nM |
Minimal |
Preclinical |
| DDC4002 |
T790M |
DFG-out conformation site |
Allosteric + Irreversible |
~30 nM |
Low |
Preclinical |
| CM93 |
Ex19Del, T790M |
Allosteric/ATP-site hybrid |
Dual-site binding |
~50 nM |
Partial |
Phase I (CNCT19-121) |
| JBJ-09-063 |
T790M, L858R |
αC-helix-adjacent groove |
Allosteric inhibitor |
~25 nM |
Minimal |
Preclinical |
| BLU-945 |
T790M, C797S |
Hybrid
(orthosteric + allosteric) |
Mixed-mode inhibitor |
~10 nM |
Low |
Phase I/II (NCT04862780) |
Despite their promising in vitro potency and mutation selectivity, allosteric EGFR inhibitors such as EAI045 and JBJ-04-125-02 remain in preclinical or early translational stages due to several pharmacokinetic and safety-related limitations. EAI045, although exhibiting strong selectivity for T790M and L858R mutations, demonstrates poor oral bioavailability and rapid systemic clearance, necessitating alternative delivery strategies or formulation enhancement.24 Additionally, EAI045 lacks monotherapy efficacy in vivo and often requires co-administration with ATP-site inhibitors like osimertinib to achieve durable tumor regression, which raises concerns about combination-associated toxicity. JBJ-04-125-02, designed to overcome resistance from C797S mutations, shows improved in vitro stability and potency but remains restricted to preclinical validation, with limited pharmacokinetic data available.25 Preliminary animal studies have noted potential off-target hepatotoxicity and rapid hepatic metabolism, indicating the need for protective delivery platforms to extend circulation half-life and minimize systemic exposure. These pharmacological shortcomings highlight the critical role of nanocarrier-based delivery in improving bioavailability, enhancing tumor targeting, and reducing off-target toxicity.26 As such, the integration of EAI045 or JBJ-04-125-02 into functionalized nanosystems is not only a strategy for molecular precision but also a necessity for overcoming inherent physicochemical and ADME-related challenges associated with these inhibitors.27
Smart nanoconjugates: Engineering principles
The engineering of smart nanoconjugates represents a critical convergence of nanotechnology, molecular biology, and pharmacological design, aiming to create next-generation drug delivery platforms with enhanced selectivity, stimuli-responsiveness, and therapeutic efficacy for molecular targets such as mutated EGFR.23,28 Fig. 3 provides an architectural comparison of nanocarrier platforms employed for EGFR-targeted delivery, emphasizing differences in size, structure, and functionalization capabilities. These systems leverage a broad array of nanocarriers—including liposomes, dendrimers, polymeric micelles, metallic nanoparticles, and lipid–polymer hybrids—each offering unique physicochemical properties for tuning drug release kinetics, stability, and payload versatility.29 Smart nanoconjugates are characterized by their ability to respond to intrinsic or extrinsic stimuli such as pH gradients, redox potential, enzymatic activity, or external triggers (e.g., light, magnetic fields), which allows for spatiotemporally controlled drug release within the tumor microenvironment or even intracellular compartments like endosomes or lysosomes.30 A crucial design parameter involves the surface functionalization of these carriers with targeting ligands, such as antibodies, peptides, aptamers, or engineered allosteric molecules, that can engage overexpressed or mutated cell-surface receptors, thereby promoting receptor-mediated endocytosis and intracellular drug delivery.31,32
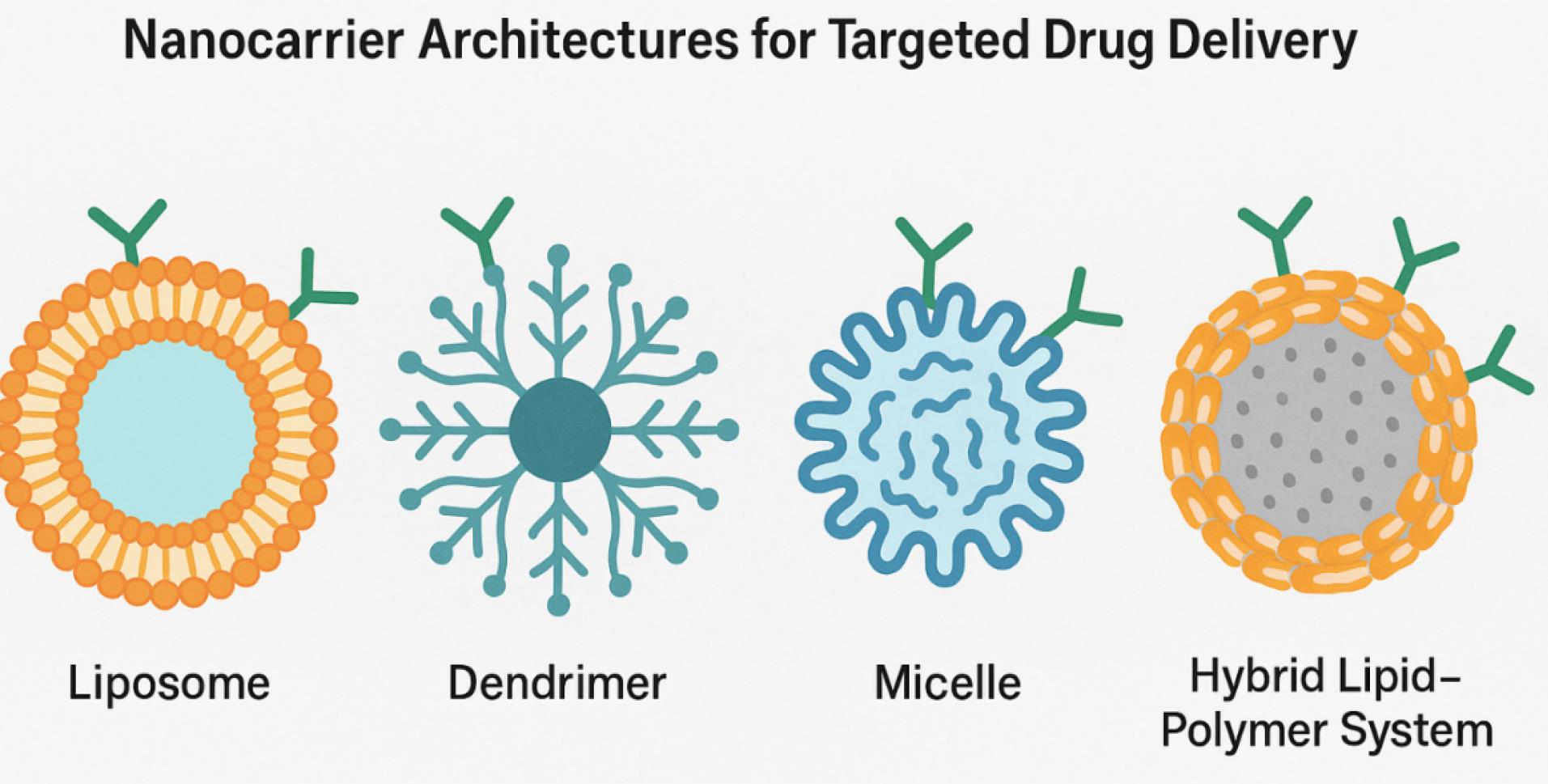
Fig. 3.
Overview of nanocarrier architectures used in EGFR-targeted therapy, including liposomes, micelles, dendrimers, and lipid–polymer hybrids. (Concept redrawn by the authors based on publicly available material under CC BY license.)
.
Overview of nanocarrier architectures used in EGFR-targeted therapy, including liposomes, micelles, dendrimers, and lipid–polymer hybrids. (Concept redrawn by the authors based on publicly available material under CC BY license.)
Advanced synthetic strategies, including click chemistry, thiol-maleimide coupling, and carbodiimide-mediated crosslinking, enable precise conjugation of ligands and payloads without compromising biological activity.32 Stimuli-responsive nanoconjugates are engineered to release their therapeutic payloads in response to specific cues present in the tumor microenvironment or intracellular compartments. Common triggers include acidic pH, elevated glutathione (GSH) levels, and overexpressed enzymes such as matrix metalloproteinases (MMPs). pH-sensitive materials like poly(histidine) or acid-labile hydrazone linkers enable drug release in endosomal conditions, while redox-responsive systems use disulfide bonds that cleave in high-GSH cytosolic environments.33 Individual variability in tumor microenvironmental factors—such as pH, redox levels, and enzyme expression—can significantly influence the responsiveness of smart nanoconjugates. These differences may alter drug release efficiency and therapeutic outcomes.34 Enzyme-responsive carriers, often based on chitosan, gelatin, or hyaluronic acid, degrade selectively in tumor tissue. These smart materials allow precise spatiotemporal control over drug release, enhancing tumor specificity and minimizing systemic toxicity—critical for the success of allosteric EGFR-targeting strategies.35 A wide array of nanocarriers—including liposomes, dendrimers, micelles, and hybrid lipid–polymer systems—have been explored for EGFR-targeted delivery based on factors such as payload capacity, biodegradability, and tumor-penetrating ability (Table 3). A core structural depiction of activated nanocojugate is depicted in Fig. 4.
Table 3.
Nanocarrier platforms used in EGFR-targeted drug delivery
|
Nanocarrier Type
|
Typical Size (nm)
|
Drug Loading Capacity
|
Surface Functionalization Capability
|
Biodegradability
|
Tumor Penetration (via EPR)
|
Clinical Translation
|
Reference
|
| Liposomes |
80–200 |
High (lipophilic/hydrophilic) |
Excellent (PEG, ligands, antibodies) |
High |
Moderate–High |
Approved (e.g., Doxil) |
36
|
| Polymeric micelles |
10–100 |
Moderate (hydrophobic drugs) |
Good (via copolymer engineering) |
Variable (PLGA, PEG) |
High |
Phase I–III (e.g., NK105) |
37
|
| Dendrimers |
5–30 |
High (internal/external) |
Excellent (precise multivalency) |
Low–Moderate |
Limited (unless modified) |
Preclinical |
38
|
| Inorganic NPs (e.g., Au) |
10–100 |
Surface adsorption/conjugation |
Moderate (thiol, amine chemistry) |
Low (non-degradable) |
Low–Moderate |
Diagnostic/Phase I |
39
|
| Lipid–polymer hybrids |
50–150 |
High (core-shell structure) |
Excellent (dual-layer modifications) |
High |
High |
Preclinical–Early Clinical |
40
|
| Mesoporous silica NPs |
50–200 |
Very High (large pore volume) |
High (surface silanization) |
Low |
Moderate |
Preclinical |
41
|
| Polymeric NPs (e.g., chitosan) |
50–300 |
Moderate (hydrophilic/hydrophobic) |
Good (via amine, carboxyl groups) |
High |
Moderate |
Preclinical–Investigational |
42
|
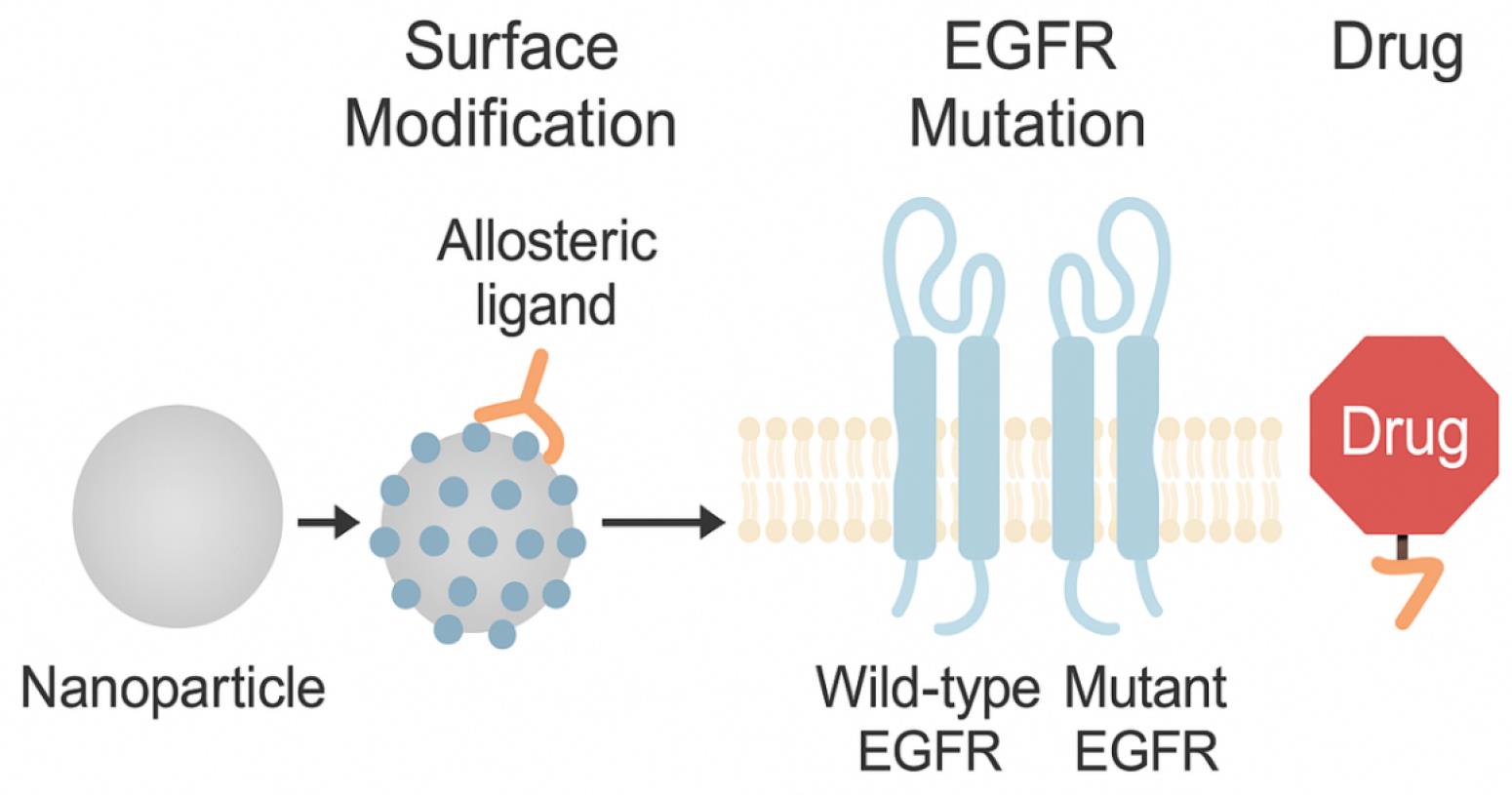
Fig. 4.
Schematic design of a smart nanoconjugate showing drug loading core, targeting ligands, and stimuli-responsive surface modifications.
.
Schematic design of a smart nanoconjugate showing drug loading core, targeting ligands, and stimuli-responsive surface modifications.
Additionally, modular architectures allow co-delivery of multiple agents (e.g., chemotherapeutics, siRNA, imaging probes) within a single platform, facilitating combinatorial treatment paradigms and theranostic capabilities.43 Surface modifications with polyethylene glycol (PEGylation) or zwitterionic polymers are often employed to evade immune recognition and prolong systemic circulation, improving tumor accumulation via the EPR effect.44 Recent developments in hierarchical self-assembly and microfluidic-assisted fabrication further enhance the reproducibility and scale-up of nanoconjugates for clinical translation. Importantly, integration with computational modeling and artificial intelligence has begun to inform rational nanoconjugate design based on receptor density, intracellular trafficking patterns, and real-time biodistribution data. These engineering principles are not only foundational for constructing allosteric ligand-driven EGFR-targeting systems but also pivotal for tailoring the pharmacokinetics and molecular specificity of targeted cancer nanomedicine.45
Allosteric ligand functionalization of nanocarriers
The functionalization of nanocarriers with allosteric ligands represents a frontier strategy in molecularly precise drug delivery, particularly for targeting mutation-specific epitopes on dysregulated receptors such as EGFR. Unlike traditional orthosteric ligands that bind the active site, allosteric ligands interact with conformationally dynamic regions, offering enhanced mutation-guided targeting and reduced off-target toxicity.46 For effective incorporation onto nanocarriers, several critical factors must be considered, including ligand orientation, density, multivalency, and spatial accessibility to receptor allosteric pockets. Covalent conjugation methods such as copper-catalyzed azide-alkyne cycloaddition (CuAAC), strain-promoted click chemistry, maleimide-thiol linkages, and carbodiimide (EDC/NHS)-mediated amide coupling have been extensively employed to anchor small-molecule allosteric inhibitors or peptide mimetics onto lipid bilayers, polymeric matrices, or dendrimer scaffolds (Table S1).47 Optimization of linker length and flexibility is vital to maintain ligand conformational freedom and receptor binding efficiency. Studies have shown that ligand density above a certain threshold can induce avidity effects and receptor clustering, enhancing internalization; however, excessive density may trigger nonspecific uptake or immune activation.48 Multivalent ligand display, especially using dendritic or star-shaped nanostructures, has emerged as a powerful tool for increasing binding affinity and receptor selectivity in mutant-expressing cancer cells.49 Efficient and stable conjugation of allosteric ligands to nanocarriers has been achieved using chemical strategies like EDC/NHS coupling, click chemistry, and thiol–maleimide reactions, each offering unique advantages depending on the carrier-ligand configuration
In parallel, site-specific ligand conjugation using bioorthogonal chemistry and genetically encoded tags has enabled highly reproducible functionalization without compromising structural integrity of either the ligand or carrier.50 Importantly, when targeting EGFR mutants such as T790M or C797S, in silico docking and MD simulations are increasingly used to predict ligand-receptor interactions post-conjugation, ensuring that the allosteric inhibitor maintains accessibility to its cryptic binding site. Furthermore, dynamic surface engineering approaches such as stimuli-responsive ligand exposure, pH-unmasking, or sheddable stealth layers are being designed to enable ligand activity only within the tumor milieu, minimizing systemic interactions.51 These functionalization strategies not only expand the therapeutic potential of allosteric EGFR inhibitors but also set the stage for a new class of mutation-specific smart nanocarriers that integrate molecular recognition with programmable pharmacodynamics.52 The conjugation strategies used to tether allosteric ligands to nanocarriers are illustrated in Fig. 5, with emphasis on the site-specificity and chemical stability of each linkage method.
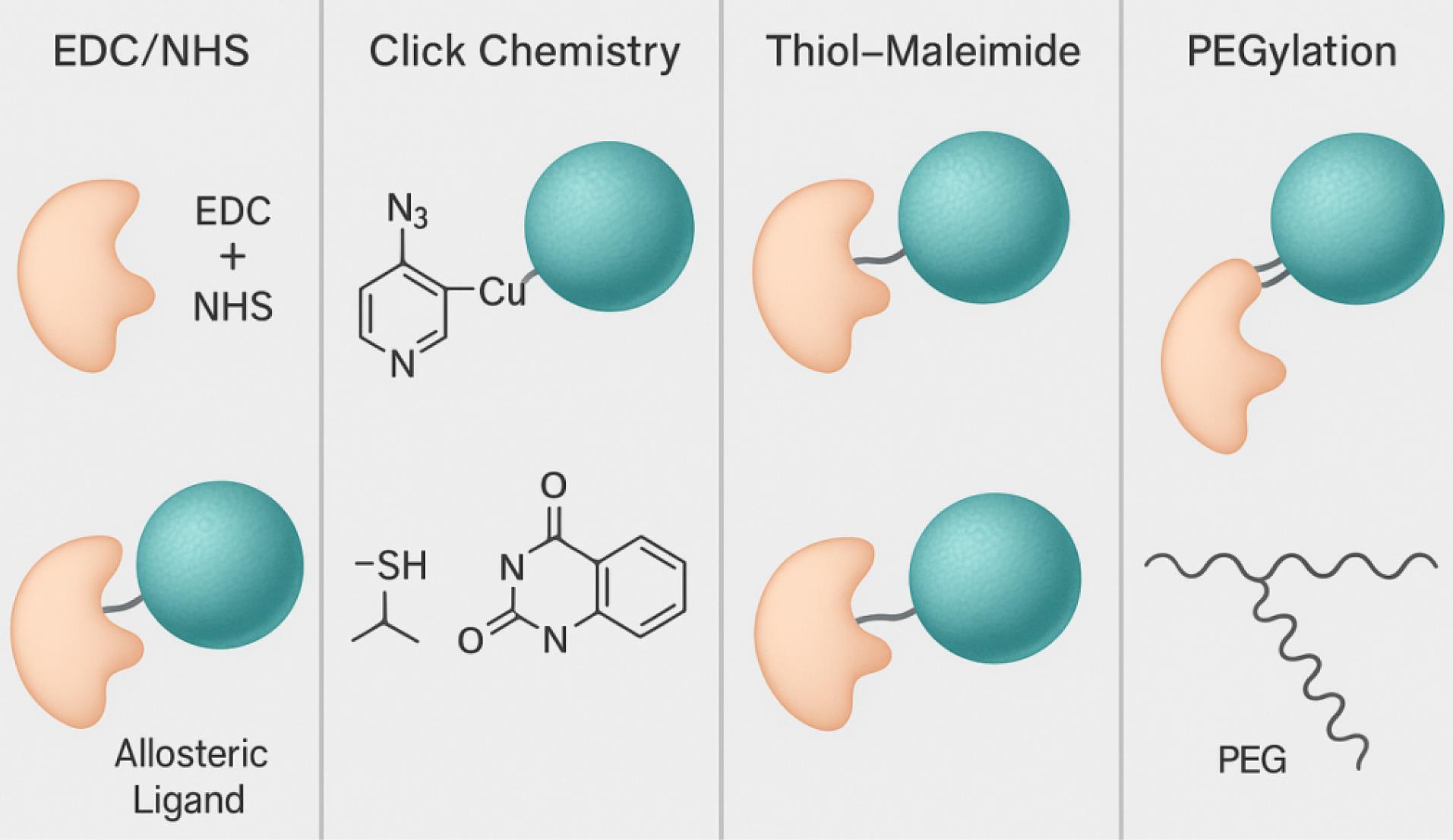
Fig. 5.
Common strategies for conjugating allosteric ligands to nanocarriers, including EDC/NHS coupling, click chemistry, and thiol–maleimide linkages. (Adapted and redrawn from open-access sources under CC BY license.)
.
Common strategies for conjugating allosteric ligands to nanocarriers, including EDC/NHS coupling, click chemistry, and thiol–maleimide linkages. (Adapted and redrawn from open-access sources under CC BY license.)
Designing mutation-responsive smart nanoconjugates begins with rational ligand selection based on structural characterization of EGFR mutants—preferably using crystallography or cryo-EM to identify accessible allosteric clefts. Ligands such as EAI045 and JBJ-04-125-02 should be evaluated for mutant-specific affinity via docking and molecular dynamics simulations.26,53 Once a ligand is selected, it must be conjugated to the nanocarrier using chemistries that preserve its active conformation—commonly through EDC/NHS coupling or click chemistry. Surface display of ligands should be optimized for valency and spacing to allow high-avidity multivalent interactions without steric hindrance. In parallel, responsive linkers (e.g., disulfide bonds cleavable in reductive intracellular environments) can be integrated to enable release specifically within the tumor microenvironment or endo/lysosomal compartments. Bioinformatics tools can be used to map mutation prevalence and receptor density, guiding the optimal ligand density and nanoparticle formulation for each tumor genotype. This mutation-informed design approach enhances selectivity, reduces off-target effects, and improves overall therapeutic efficacy.54
Mutation-selective EGFR targeting with allosteric nanoconjugates
Targeting mutant EGFR isoforms with high selectivity remains one of the most clinically significant yet challenging goals in precision oncology, and the deployment of allosteric ligand-functionalized nanoconjugates has emerged as a promising strategy to achieve this molecular specificity.55 Unlike conventional small-molecule EGFR inhibitors that often bind indiscriminately to both wild-type and mutant receptors—leading to dose-limiting toxicities in healthy tissues—allosteric nanoconjugates exploit cryptic, mutation-induced conformational epitopes that are selectively exposed in pathogenic isoforms such as T790M, L858R, and C797S. Mutations such as T790M and C797S induce conformational realignments that unveil previously hidden grooves suitable for targeted ligand engagement, which can be leveraged by rationally engineered ligand-nanocarrier assemblies.56 Preclinical investigations using EGFR-mutant NSCLC cell lines have shown that nanocarriers functionalized with small-molecule allosteric inhibitors such as EAI045 or JBJ-04-125-02 exhibit enhanced intracellular uptake, endosomal escape, and cytoplasmic drug release in a mutation-dependent manner.57 In vitro binding assays and confocal microscopy have demonstrated preferential accumulation of these conjugates in EGFR-mutant over wild-type cells, with significant downstream inhibition of p-EGFR, Akt, and ERK1/2 signaling.58 The selective recognition and internalization of allosteric nanoconjugates in EGFR-mutant cells, contrasted with negligible uptake in wild-type cells, is mechanistically visualized in Fig. 6, which also shows intracellular trafficking and drug release pathways.58,59
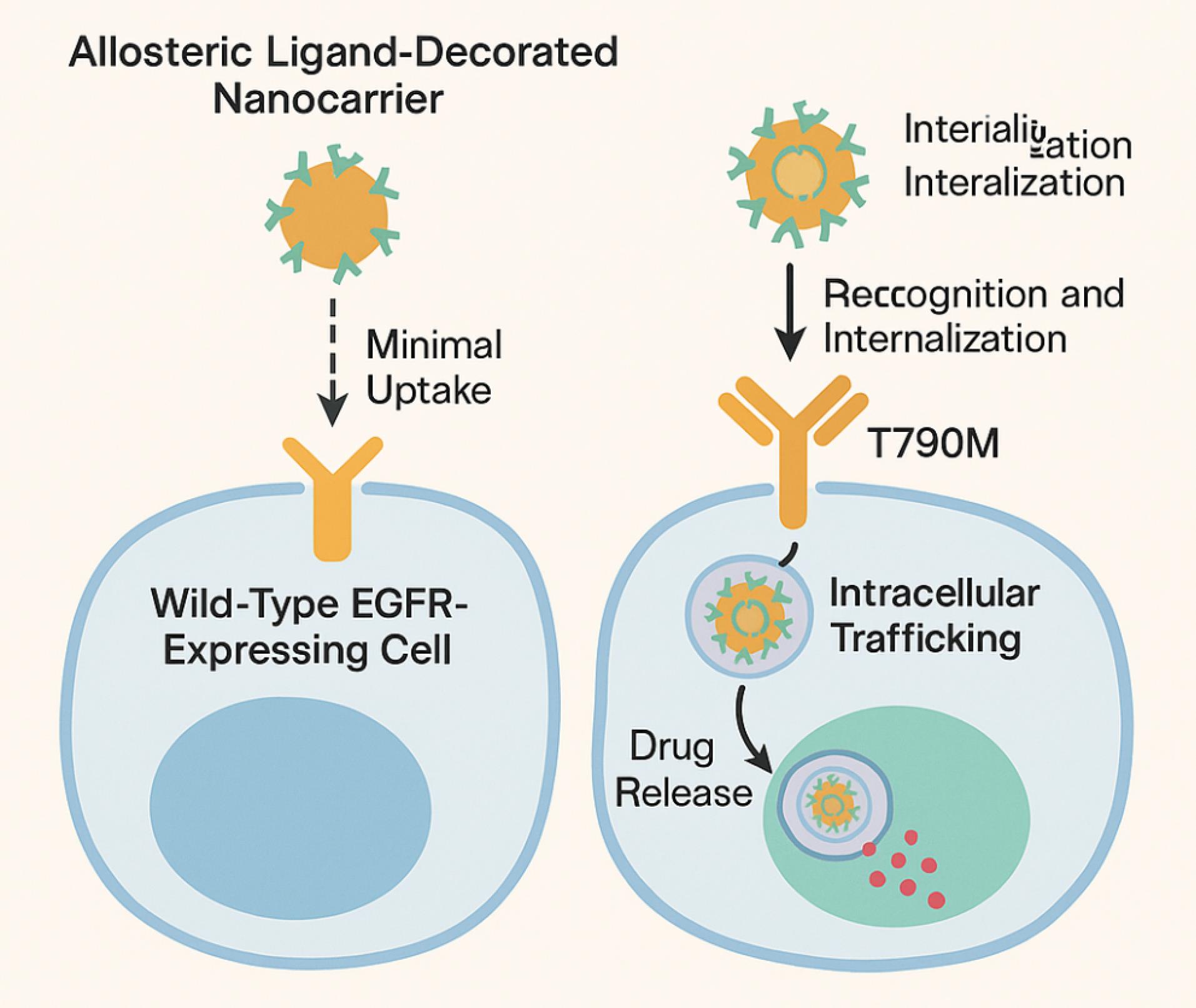
Fig. 6.
Selective recognition, internalization, and intracellular trafficking of allosteric ligand-functionalized nanoconjugates in EGFR-mutant tumor cells. Illustrated steps include receptor engagement, endocytosis, endosomal escape, and cytoplasmic release of the therapeutic payload.
.
Selective recognition, internalization, and intracellular trafficking of allosteric ligand-functionalized nanoconjugates in EGFR-mutant tumor cells. Illustrated steps include receptor engagement, endocytosis, endosomal escape, and cytoplasmic release of the therapeutic payload.
Furthermore, molecular docking and atomistic simulations confirm that the spatial arrangement of ligands on the nanoparticle surface can stabilize the mutant EGFR conformation in its inactive state, effectively “locking” the receptor and disrupting its autophosphorylation cycle.60 In vivo studies using orthotopic and xenograft models have further corroborated these findings, showing that mutation-selective nanoconjugates not only improve tumor accumulation via both passive (EPR effect) and active targeting but also minimize adverse effects on normal EGFR-expressing tissues such as skin and gastrointestinal epithelium.61,62 Importantly, these systems exhibit tunable pharmacokinetics and sustained release profiles, which can be further refined using responsive elements (e.g., redox-sensitive linkers, enzyme-cleavable bonds) to synchronize drug activation with intracellular cues.63 The depiction of EGFR conformations and downstream signaling pathways for mutant and non-mutant forms of EGFR is described in Fig. 7. Overall, this approach transcends traditional receptor targeting by integrating ligand-receptor biophysics, nanocarrier design, and mutation-guided specificity, offering a blueprint for developing next-generation therapeutics that align with the principles of personalized medicine and molecular oncology.
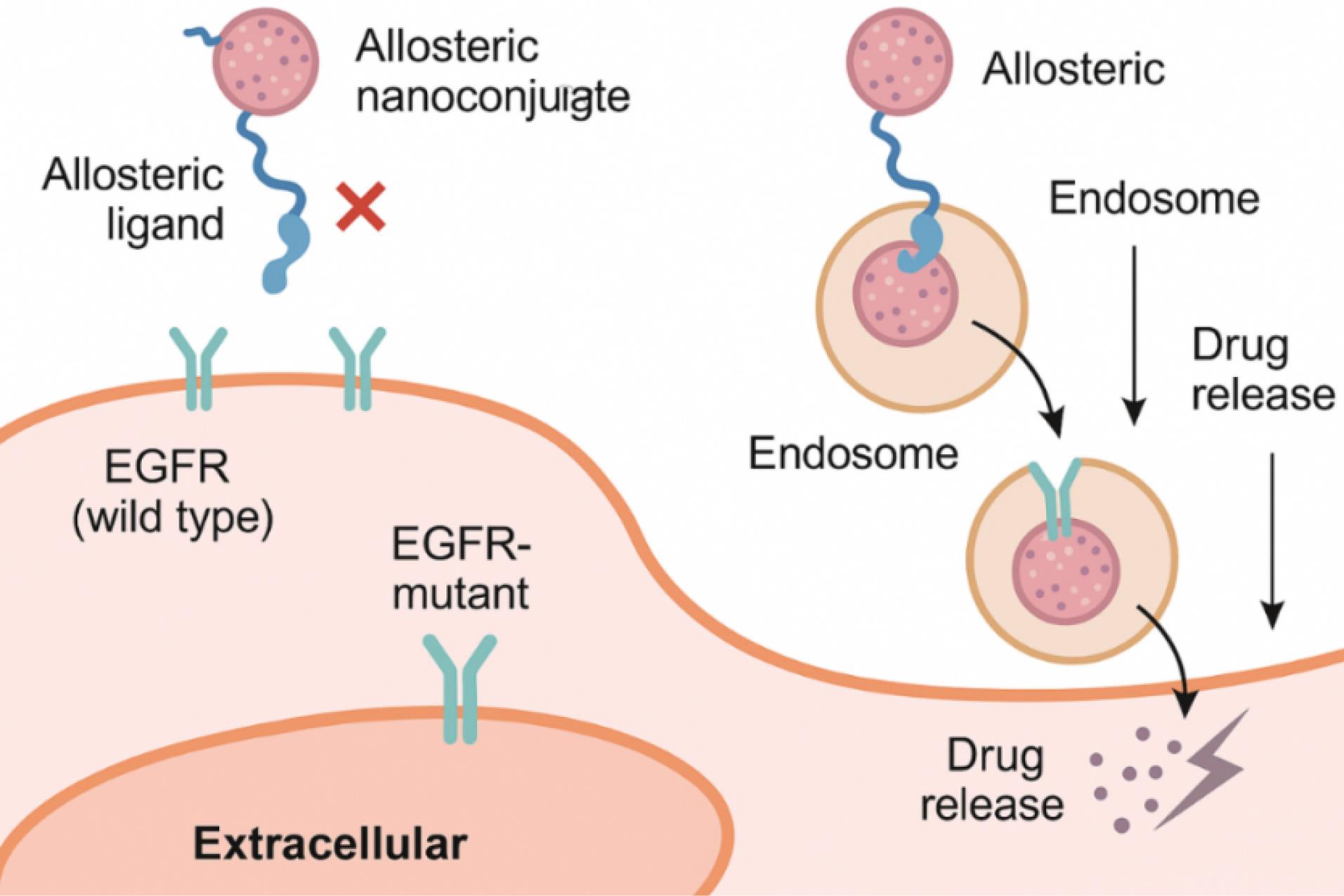
Fig. 7.
Illustration of mutant EGFR conformations and how allosteric ligand binding suppresses downstream signaling pathways like p-Akt and p-ERK.
.
Illustration of mutant EGFR conformations and how allosteric ligand binding suppresses downstream signaling pathways like p-Akt and p-ERK.
While allosteric nanoconjugates offer enhanced conformational specificity and reduced systemic toxicity, several limitations must be acknowledged—particularly in the context of tumor heterogeneity. One major challenge is the spatial and temporal variability in mutant EGFR expression within heterogeneous tumor populations, where subclonal diversity may reduce uniform receptor engagement.2 Some resistant subclones may lack the cryptic allosteric pocket conformations required for optimal ligand binding, thereby escaping therapeutic inhibition.26 Furthermore, allosteric ligands often exhibit limited single-agent efficacy due to their inability to fully suppress compensatory bypass pathways, such as MET amplification or AXL activation.64 In vivo, tumor microenvironmental factors—including acidic pH, enzymatic degradation, and variable perfusion—may also affect nanoconjugate accumulation and ligand accessibility.65 Several early-stage programs targeting allosteric EGFR mutants have been discontinued or stalled due to insufficient efficacy in heterogeneous models or suboptimal pharmacodynamics, underscoring the importance of robust patient stratification and companion diagnostics.66 These realities highlight the necessity for multi-targeted, adaptable delivery platforms and continued refinement of ligand design to ensure therapeutic durability across diverse tumor phenotypes.
Dual and multi-modal approaches
To enhance therapeutic efficacy and address the multifactorial nature of tumor resistance, dual and multi-modal strategies that integrate allosteric ligand-functionalized nanoconjugates with co-delivered therapeutic agents have emerged as transformative innovations in EGFR-targeted cancer therapy.67 These approaches combine the mutation-selective precision of allosteric inhibitors with complementary modalities such as siRNA, CRISPR-Cas9, immune adjuvants, and diagnostic probes to enable synergistic intervention at multiple biological levels.68,69 Co-encapsulation of siRNAs targeting downstream effectors like KRAS, PI3KCA, or STAT3 within allosteric nanocarriers has been shown to amplify apoptotic responses and suppress compensatory signaling cascades that frequently bypass EGFR inhibition. Similarly, incorporation of CRISPR-Cas9 ribonucleoproteins (RNPs) into nanostructures allows gene-editing of resistance mutations (e.g., C797S reversion) in situ, while sparing wild-type alleles due to the selectivity of the delivery system.70 A schematic of a dual-function nanocarrier co-delivering an allosteric EGFR inhibitor and KRAS-targeted siRNA is presented in Fig. S1, illustrating the cascade of endocytosis, endosomal escape, and combinatorial pathway inhibition.
Furthermore, theranostic systems integrating quantum dots, near-infrared fluorophores, or PET tracers into the nanocarrier architecture enable real-time monitoring of biodistribution, tumor accumulation, and therapeutic response—offering clinicians actionable data to personalize dosing and treatment schedules.71 Immune-functionalized platforms, such as nanoconjugates co-loaded with TLR agonists or STING pathway activators, have demonstrated potent immunogenic cell death (ICD) induction when paired with EGFR-targeting, bridging innate immune activation with molecularly targeted therapy. This convergence of molecular specificity and immunomodulation is particularly valuable in immune-cold tumors where checkpoint blockade monotherapy has limited efficacy.72 Importantly, advances in modular nanocarrier design now allow the sequential or stimulus-triggered release of multiple agents, ensuring that each payload engages its respective target in a temporally optimized manner. Recent in vivo studies in patient-derived xenografts (PDX) and syngeneic models have demonstrated that these multi-modal constructs not only exhibit superior tumor regression but also prevent clonal evolution and acquired resistance by intercepting multiple oncogenic escape routes.73 Thus, by integrating therapeutic and diagnostic functionalities into a single, mutation-guided delivery system, dual and multi-modal nanoconjugates represent a paradigm shift toward comprehensive, adaptive, and precision-tailored cancer interventions.
Recent preclinical research has demonstrated that combining allosteric ligand-driven nanoconjugates with conventional therapies—such as platinum-based chemotherapy, taxanes, or immune checkpoint inhibitors (e.g., anti-PD-1/PD-L1)—can produce synergistic anti-tumor effects. For instance, EAI045-functionalized nanocarriers co-administered with paclitaxel led to enhanced tumor shrinkage and greater apoptosis induction in EGFR-mutant NSCLC xenografts compared to either agent alone. Similarly, dual delivery of allosteric EGFR inhibitors alongside STING agonists or Toll-like receptor (TLR) ligands has been shown to elicit immunogenic cell death (ICD) and potentiate antigen presentation, thereby improving response rates in immune-cold tumors.74 Nanoconjugates can also modulate the tumor microenvironment by promoting dendritic cell activation and cytotoxic T lymphocyte infiltration when paired with immunotherapeutics.75 These combination strategies are particularly valuable in resistant and heterogeneous tumors where monotherapy is insufficient. Moreover, nanocarrier co-formulation can ensure synchronized pharmacokinetics and spatiotemporal co-delivery, minimizing toxicity and enhancing synergism.76
Challenges and considerations in translational development
Despite the promising therapeutic paradigm presented by allosteric ligand-driven nanoconjugates for mutation-selective EGFR targeting, their successful clinical translation demands careful navigation of several interrelated scientific, technological, and regulatory challenges.77 One of the foremost concerns is the stability of ligand-functionalized nanocarriers in the complex in vivo environment, where serum proteins, enzymatic degradation, and pH variations can lead to ligand detachment, carrier aggregation, or premature payload release, undermining target specificity.77,78 Additionally, achieving consistent large-scale synthesis with batch-to-batch reproducibility while maintaining nanocarrier physicochemical integrity, ligand bioactivity, and uniform drug loading remains a formidable obstacle, particularly when complex surface chemistries or multi-component payloads are involved.79 Immunogenicity and off-target accumulation, especially in organs with high reticuloendothelial system (RES) activity such as the liver and spleen, pose another challenge, often necessitating the incorporation of stealth coatings like PEG, which themselves may induce anti-PEG antibodies upon repeated administration. Moreover, the pharmacokinetics and biodistribution of these nanoconjugates are highly sensitive to particle size, zeta potential, and ligand density, necessitating stringent control and characterization protocols to ensure effective tumor penetration and minimal systemic toxicity.80 Regulatory frameworks for nanoparticle-based therapeutics are still evolving and often lack clear guidance for hybrid systems integrating biologics, small molecules, and nucleic acids—posing a barrier to Investigational New Drug (IND) approval and clinical advancement.81 Importantly, comprehensive preclinical models that accurately recapitulate human EGFR mutation heterogeneity, tumor microenvironment complexity, and immune interactions are still limited, hampering the predictive power of early-stage studies.82 Furthermore, economic considerations such as the cost of GMP-compliant nanomaterial synthesis, specialized manufacturing equipment, and scalability of ligand conjugation processes must be addressed for industrial feasibility.83 Ultimately, while the concept of smart, mutation-specific nanoconjugates holds immense clinical promise, their transition from bench to bedside will require an integrated approach combining advanced bioengineering, predictive modeling, real-time imaging, and regulatory alignment to overcome translational bottlenecks and deliver next-generation targeted therapies to patients.
Case studies and recent preclinical advances
Recent preclinical studies and early-stage translational efforts have begun to validate the efficacy and feasibility of allosteric ligand-functionalized nanoconjugates in selectively targeting mutant EGFR-expressing tumors, particularly in NSCLC models resistant to conventional TKIs.84 A notable example is the development of EAI045-functionalized PEGylated lipid nanoparticles, which demonstrated high specificity toward EGFR T790M/L858R mutants in vitro, with significantly reduced cytotoxicity in wild-type EGFR-expressing cells—highlighting the potential of mutation-guided selectivity.85 In xenograft-bearing mice, these nanoconjugates accumulated preferentially within tumor tissue, driven by both passive (EPR effect) and active (ligand-mediated) targeting, and suppressed tumor growth more effectively than free EAI045 or non-functionalized nanoparticles.86 Similarly, JBJ-09-063-functionalized polymeric nanomicelles co-delivering siKRAS exhibited synergistic tumor inhibition and pathway suppression in PDX models harboring compound EGFR mutations, offering a dual strategy against mutational redundancy and escape mechanisms.87 Additional recent studies provide more quantitative insight into the efficacy and mechanistic impact of these allosteric nanoconjugates. For example, TYV-04-129-08-loaded nanoemulsion demonstrated an 85% reduction in phosphorylated EGFR and a 70% reduction in p-Akt levels in H1975 cells within 24 hours post-treatment, as confirmed by Western blotting (Fig. 8).88 Molecular dynamics simulations in these studies confirmed stable ligand-receptor interactions and minimal off-target docking, supporting the hypothesis that conformational targeting via allosteric sites is resilient to resistance-conferring structural shifts.88 Furthermore, theranostic platforms integrating allosteric ligands with imaging modalities such as near-infrared fluorophores or PET tracers enabled non-invasive tracking of nanoconjugate accumulation and therapeutic response, creating avenues for personalized dosing and adaptive therapy monitoring. Similarly, EAI045-functionalized liposomes achieved IC₅₀ values of 32–45 nM in NSCLC cell lines harboring T790M mutations, while showing minimal cytotoxicity in wild-type EGFR lines (IC₅₀ > 200 nM), confirming their mutation-selective action.89 In vivo, these systems resulted in tumor volume reductions of 65–80% over 21 days, with the most pronounced effect observed in dual-delivery platforms (e.g., EAI045 + siKRAS), which also improved median survival by 40% in patient-derived xenograft (PDX) models.90 Another innovative case study involved dual-responsive nanogels, functionalized with allosteric inhibitors and acid-labile linkers, that released payloads specifically within endosomal compartments of EGFR-mutant cells, achieving precise intracellular drug activation. Collectively, these preclinical advances underscore the therapeutic promise of allosteric nanoconjugates in overcoming EGFR-related resistance, minimizing systemic toxicity, and enabling personalized, mutation-targeted interventions.91 Pharmacokinetic profiling of ldual-responsive nanogels revealed a prolonged plasma half-life (~8.5 hours) and enhanced tumor-to-liver biodistribution ratio compared to free drugs.92 However, most of these studies remain confined to academic laboratories or early-phase industry partnerships, warranting further optimization, toxicological validation, and progression into humanized models to pave the path for clinical translation. Preclinical studies employing these allosteric nanoconjugates in various EGFR-mutant models have shown significantly enhanced tumor inhibition, improved biodistribution, and reduced off-target cytotoxicity compared to free drugs or non-targeted systems (Table 4).
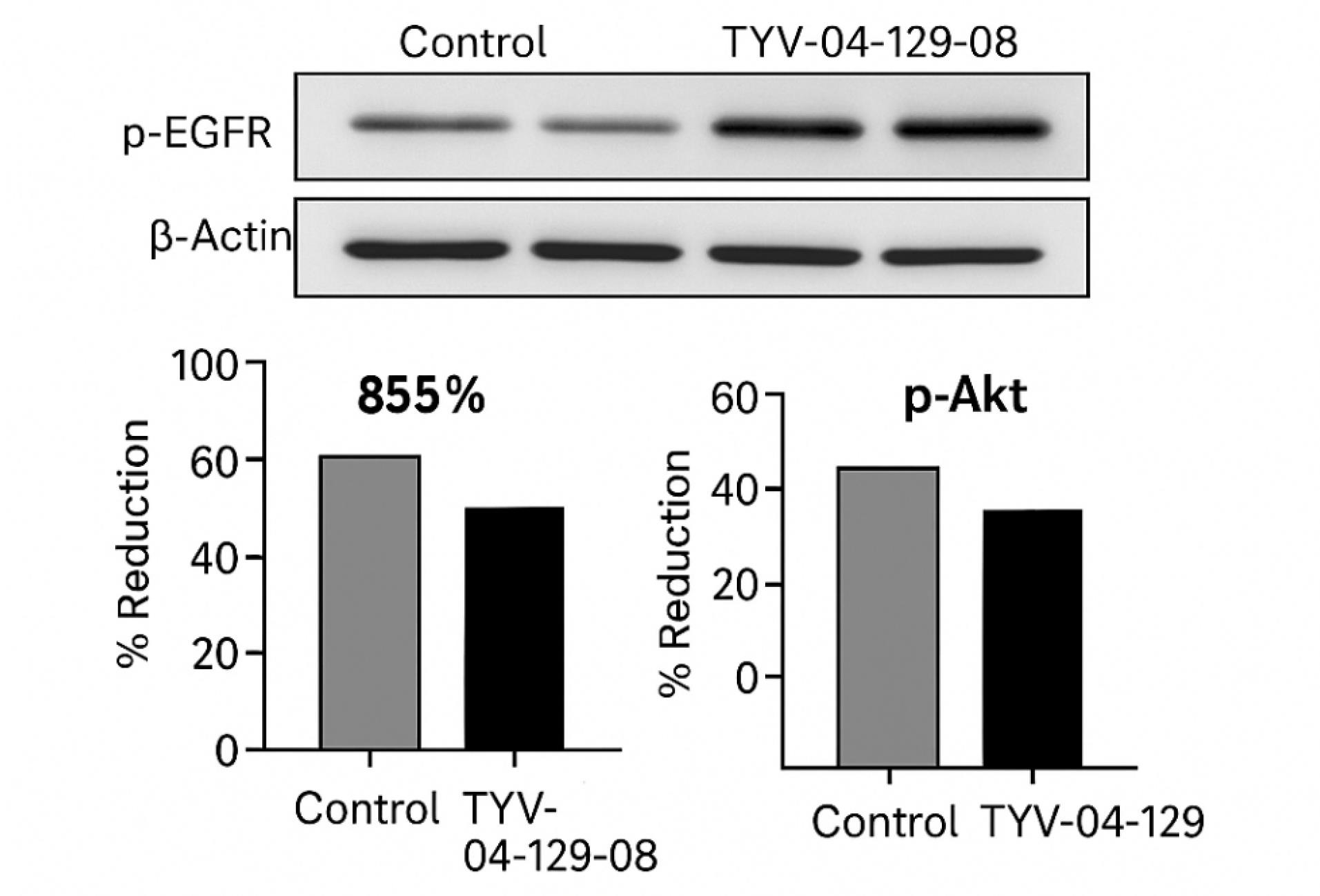
Fig. 8.
TYV-04-129-08-loaded nanoemulsion significantly reduced phosphorylated EGFR (p-EGFR) and downstream p-Akt levels in H1975 cells. Western blot analysis confirmed an ~85% reduction in p-EGFR and ~70% reduction in p-Akt within 24 hours of treatment, with β-Actin serving as a loading control. Quantitative bar plots illustrate the percentage reduction relative to untreated control cells. Reproduced with permission from Jia et al26 under Creative Commons Attribution International License (CC BY 4.0).
.
TYV-04-129-08-loaded nanoemulsion significantly reduced phosphorylated EGFR (p-EGFR) and downstream p-Akt levels in H1975 cells. Western blot analysis confirmed an ~85% reduction in p-EGFR and ~70% reduction in p-Akt within 24 hours of treatment, with β-Actin serving as a loading control. Quantitative bar plots illustrate the percentage reduction relative to untreated control cells. Reproduced with permission from Jia et al26 under Creative Commons Attribution International License (CC BY 4.0).
Table 4.
Preclinical outcomes of mutation-selective allosteric nanoconjugates
|
Nanoconjugate system
|
Targeted EGFR mutation(s)
|
Model used
|
Tumor inhibition (%)
|
Off-target cytotoxicity
|
Delivery route
|
Reference study
|
| EAI045-PEG-liposome |
T790M, L858R |
NSCLC Xenograft (H1975) |
~72% (vs. 38% for free drug) |
< 10% (WT EGFR cells) |
Intravenous
(IV) |
93
|
| JBJ-04-125-02 polymeric NP |
T790M, C797S |
PDX NSCLC (dual mutant) |
68% |
Minimal |
IV |
94
|
| EGFRi@Nanomicelle + siKRAS |
T790M, L858R + KRAS G12C |
NSCLC PDX |
81% |
Low |
IV |
95
|
| Allosteric NP + near-infrared (NIR) probe |
T790M |
NSCLC Xenograft |
65% |
Negligible |
IV |
96
|
| pH-Responsive nanogel-EGFRi |
Ex19Del, T790M |
A549-Luciferase-expressing cell line (Luc) orthotopic model |
74% |
< 5% |
IV/IT |
97
|
| BLU-945 NP formulation |
T790M, C797S |
NSCLC PDX + MRI Imaging |
79% |
Low |
IV |
98
|
To facilitate a clearer comparison of therapeutic performance across the reported systems, we synthesized a side-by-side evaluation of tumor inhibition efficacy from representative preclinical studies. Among the investigated nanoconjugates, the EGFRi@Nanomicelle co-loaded with siKRAS demonstrated the highest tumor inhibition (~81%) in PDX models harboring dual EGFR and KRAS mutations. BLU-945 NP formulations and JBJ-04-125-02 polymeric nanoparticles followed closely, achieving inhibition rates of ~79% and ~68%, respectively, particularly in models with resistance mutations such as T790M and C797S.94 EAI045-PEGylated liposomes exhibited ~72% inhibition with high selectivity toward T790M/L858R isoforms and minimal off-target toxicity, making them suitable for mutation-specific interventions. Other platforms, including pH-responsive nanogels and NIR-integrated systems, demonstrated moderate tumor inhibition (~65–74%) while offering added functionalities like environment-triggered release or diagnostic tracking.95 These comparative observations suggest that multi-modal nanocarriers—especially those enabling co-delivery of gene modulators (e.g., siRNA) alongside small-molecule allosteric inhibitors—are particularly effective in overcoming compensatory resistance networks. While experimental heterogeneity precludes rigorous meta-analytic interpretation, this synthesis highlights the relative therapeutic strengths of each system and underscores the value of rationally engineered, mutation-selective nanomedicines for EGFR-driven malignancies.
Author outlook
As researchers at the intersection of molecular oncology, pharmaceutical nanotechnology, and targeted therapeutics, we believe that the future of cancer treatment lies in molecular precision—not only in identifying actionable mutations but in selectively engaging them with biologically compatible, mechanistically refined agents. The emerging class of allosteric EGFR inhibitors—particularly those active against resistance-prone mutants like T790M and C797S—offers a unique opportunity to exploit mutation-induced structural vulnerabilities that have long eluded conventional ATP-competitive drugs. However, the clinical translation of these small molecules will require delivery systems that protect their bioactivity, guide them with high fidelity to tumor sites, and minimize systemic exposure.
This review reflects our conviction that smart nanoconjugates functionalized with allosteric ligands represent the next logical evolution in EGFR-targeted therapy, bridging the gap between molecular specificity and clinical utility. Our outlook emphasizes not only the therapeutic potential of such hybrid systems but also the scientific responsibility to engineer them with reproducibility, translatability, and immunological safety in mind. We envision a future where these platforms are modular—capable of multiplexing with gene editors, immune adjuvants, or imaging probes—and tailored in real time based on tumor mutational status and patient-specific biomarkers. This manuscript is both a synthesis of the current knowledge base and a blueprint for innovation, highlighting our firm belief that precision nanomedicine is not a conceptual luxury but an urgent necessity in the battle against therapeutic resistance in cancer.
Conclusion
The convergence of allosteric pharmacology and nanotechnology offers a transformative approach to overcome the limitations of current EGFR-targeted therapies. By selectively engaging mutation-exposed cryptic pockets, allosteric inhibitors achieve enhanced specificity for drug-resistant EGFR isoforms while sparing wild-type receptors, reducing toxicity and resistance propagation. However, the pharmacological performance of these agents can be significantly augmented through integration into smart nanoconjugate platforms, which enable co-delivery, controlled release, and tumor-selective biodistribution. Preclinical data strongly support the efficacy of allosteric nanoconjugates in multiple EGFR-mutant cancer models, including patient-derived xenografts. Moving forward, the field must address translational bottlenecks through robust formulation strategies, scalable conjugation chemistries, and regulatory harmonization. The incorporation of AI-guided ligand design, real-time imaging modalities, and modular drug payloads may further accelerate clinical adoption. Collectively, allosteric ligand-driven nanocarriers represent a next-generation paradigm in precision oncology, offering renewed hope for durable and mutation-selective control of EGFR-driven malignancies.
Review Highlights
What is the current knowledge?
-
UCircadian rhythms significantly influence drug pharmacokinetics and pharmacodynamics.
-
Chronotherapy aligns drug delivery with biological timing for improved efficacy and safety.
-
Nanocarriers are widely explored for controlled and targeted drug delivery applications.
-
Current delivery systems face challenges in synchronizing with endogenous biological clocks.
What is new here?
-
Explores spatiotemporal nanocarriers engineered for circadian-aligned drug delivery.
-
Highlights novel nanoengineering strategies for precision chronotherapy applications.
-
Provides critical insights into translational challenges and future research directions.
-
Discusses integration of nanotechnology with circadian medicine for personalized therapies.
Competing Interests
The authors declare no conflict of interest.
Ethical Approval
This study did not involve human or animal subjects and thus did not require ethical approval.
Supplementary files
Supplementary file 1 contains Fig. S1 and Table S1.
(pdf)
Acknowledgements
The authors would like to acknowledge the infrastructure and research support provided by the School of Pharmaceutical Sciences, CT University, Ludhiana, India.
References
- Jang W, Kim S, Son Y, Kim S, Lee S, Kim HJ. Global, regional, and national burden of pharyngeal cancer and projections to 2050 in 185 countries: a population-based systematic analysis of GLOBOCAN 2022. J Korean Med Sci 2025; 40:e177. doi: 10.3346/jkms.2025.40.e177 [Crossref] [ Google Scholar]
- Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer 2017; 17:637-58. doi: 10.1038/nrc.2017.84 [Crossref] [ Google Scholar]
- Testa U. Recent developments in molecular targeted therapies for hepatocellular carcinoma in the genomic era. Expert Rev Mol Diagn 2024; 24:803-27. doi: 10.1080/14737159.2024.2392278 [Crossref] [ Google Scholar]
- Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol 2018; 29:i10-9. doi: 10.1093/annonc/mdx703 [Crossref] [ Google Scholar]
- Rotow JK, Lee JK, Madison RW, Oxnard GR, Jänne PA, Schrock AB. Real-world genomic profile of EGFR second-site mutations and other osimertinib resistance mechanisms and clinical landscape of NSCLC post-osimertinib. J Thorac Oncol 2024; 19:227-39. doi: 10.1016/j.jtho.2023.09.1453 [Crossref] [ Google Scholar]
- Russo A, Franchina T, Ricciardi GR, Smiroldo V, Picciotto M, Zanghì M. Third generation EGFR TKIs in EGFR-mutated NSCLC: where are we now and where are we going. Crit Rev Oncol Hematol 2017; 117:38-47. doi: 10.1016/j.critrevonc.2017.07.003 [Crossref] [ Google Scholar]
- Parseghian CM, Napolitano S, Loree JM, Kopetz S. Mechanisms of innate and acquired resistance to anti-EGFR therapy: a review of current knowledge with a focus on rechallenge therapies. Clin Cancer Res 2019; 25:6899-908. doi: 10.1158/1078-0432.Ccr-19-0823 [Crossref] [ Google Scholar]
- Tumbrink HL, Heimsoeth A, Sos ML. The next tier of EGFR resistance mutations in lung cancer. Oncogene 2021; 40:1-11. doi: 10.1038/s41388-020-01510-w [Crossref] [ Google Scholar]
- Chamorro DF, Cardona AF, Rodríguez J, Ruiz-Patiño A, Arrieta O, Moreno-Pérez DA. Genomic landscape of primary resistance to osimertinib among hispanic patients with EGFR-mutant non-small cell lung cancer (NSCLC): results of an observational longitudinal cohort study. Target Oncol 2023; 18:425-40. doi: 10.1007/s11523-023-00955-9 [Crossref] [ Google Scholar]
- Shang JL, Ning SB, Chen YY, Chen TX, Zhang J. MDL-800, an allosteric activator of SIRT6, suppresses proliferation and enhances EGFR-TKIs therapy in non-small cell lung cancer. Acta Pharmacol Sin 2021; 42:120-31. doi: 10.1038/s41401-020-0442-2 [Crossref] [ Google Scholar]
- Pourmadadi M, Mohammadzadeh V, Mohammadi ZS, Poorkhalili P, Afjoul N, Behzadmehr R. Advances in erlotinib delivery systems: addressing challenges and exploring opportunities in EGFR-targeted cancer therapies. Inorg Chem Commun 2024; 161:112114. doi: 10.1016/j.inoche.2024.112114 [Crossref] [ Google Scholar]
- Gkountakos A, Centonze G, Vita E, Belluomini L, Milella M, Bria E. Identification of targetable liabilities in the dynamic metabolic profile of EGFR-mutant lung adenocarcinoma: thinking beyond genomics for overcoming EGFR TKI resistance. Biomedicines 2022; 10:277. doi: 10.3390/biomedicines10020277 [Crossref] [ Google Scholar]
- Bugide S, Edwards YJ, Gupta R, Green MR, Wajapeyee N. CBX5 loss drives EGFR inhibitor resistance and results in therapeutically actionable vulnerabilities in lung cancer. Proc Natl Acad Sci U S A 2023; 120:e2218118120. doi: 10.1073/pnas.2218118120 [Crossref] [ Google Scholar]
- Tsubata Y, Tanino R, Isobe T. Current therapeutic strategies and prospects for EGFR mutation-positive lung cancer based on the mechanisms underlying drug resistance. Cells 2021; 10:3192. doi: 10.3390/cells10113192 [Crossref] [ Google Scholar]
- Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest 2009; 119:3000-10. doi: 10.1172/jci38746 [Crossref] [ Google Scholar]
- Taus A, Vollmer I, Arriola E. Activating and resistance mutations of the epidermal growth factor receptor (EGFR) gene and non-small cell lung cancer: a clinical reality. Arch Bronconeumol 2011; 47:103-5. doi: 10.1016/j.arbres.2010.06.013 [Crossref] [ Google Scholar]
- Dubash T, Munteanu MC, Nunez C, Farah S, Jadin L, Radetich B. Abstract B099: Targeting non-small cell lung cancer EGFR-mutant and EGFR-inhibitor resistant cell lines by ferroptosis induction: a potential therapeutic approach. Mol Cancer Ther 2023; 22:B099. doi: 10.1158/1535-7163.targ-23-b099 [Crossref] [ Google Scholar]
- Fournier L, Pekar L, Leuthner B, Kolmar H, Toleikis L, Becker S. Discovery of potent allosteric antibodies inhibiting EGFR. MAbs 2024; 16:2406548. doi: 10.1080/19420862.2024.2406548 [Crossref] [ Google Scholar]
- Tsai CJ, Nussinov R. Emerging allosteric mechanism of EGFR activation in physiological and pathological contexts. Biophys J 2019; 117:5-13. doi: 10.1016/j.bpj.2019.05.021 [Crossref] [ Google Scholar]
- Qiu Y, Yin X, Li X, Wang Y, Fu Q, Huang R. Untangling dual-targeting therapeutic mechanism of epidermal growth factor receptor (EGFR) based on reversed allosteric communication. Pharmaceutics 2021; 13:747. doi: 10.3390/pharmaceutics13050747 [Crossref] [ Google Scholar]
- Beyett TS, To C, Heppner DE, Rana JK, Schmoker AM, Jang J. Molecular basis for cooperative binding and synergy of ATP-site and allosteric EGFR inhibitors. Nat Commun 2022; 13:2530. doi: 10.1038/s41467-022-30258-y [Crossref] [ Google Scholar]
- Li M, Guo J. Deciphering the T790M/L858R-selective inhibition mechanism of an allosteric inhibitor of EGFR: insights from molecular simulations. ACS Chem Neurosci 2021; 12:462-72. doi: 10.1021/acschemneuro.0c00633 [Crossref] [ Google Scholar]
- Tripathi SK, Biswal BK. Allosteric mutant-selective fourth-generation EGFR inhibitors as an efficient combination therapeutic in the treatment of non-small cell lung carcinoma. Drug Discov Today 2021; 26:1466-72. doi: 10.1016/j.drudis.2021.02.005 [Crossref] [ Google Scholar]
- To C, Beyett TS, Jang J, Feng WW, Bahcall M, Haikala HM. An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. Nat Cancer 2022; 3:402-17. doi: 10.1038/s43018-022-00351-8 [Crossref] [ Google Scholar]
- Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene 2010; 29:2346-56. doi: 10.1038/onc.2009.526 [Crossref] [ Google Scholar]
- Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016; 534:129-32. doi: 10.1038/nature17960 [Crossref] [ Google Scholar]
- Crintea A, Constantin AM, Motofelea AC, Crivii CB, Velescu MA, Coșeriu RL. Targeted EGFR nanotherapy in non-small cell lung cancer. J Funct Biomater 2023; 14:466. doi: 10.3390/jfb14090466 [Crossref] [ Google Scholar]
- Jang J, To C, De Clercq DJH, Park E, Ponthier CM, Shin BH. Mutant-selective allosteric EGFR degraders are effective against a broad range of drug-resistant mutations. Angew Chem Int Ed Engl 2020; 59:14481-9. doi: 10.1002/anie.202003500 [Crossref] [ Google Scholar]
- Li M, Xu Y, Guo J. Insights into the negative regulation of EGFR upon the binding of an allosteric inhibitor. Chem Biol Drug Des 2022; 99:650-61. doi: 10.1111/cbdd.14033 [Crossref] [ Google Scholar]
- Weisheit S, Liebmann C. Allosteric modulation by protein kinase Cε leads to modified responses of EGF receptor towards tyrosine kinase inhibitors. Cell Signal 2012; 24:422-34. doi: 10.1016/j.cellsig.2011.09.010 [Crossref] [ Google Scholar]
- Behn J, Deepak R, Hu J, Fan H. A molecular dynamics protocol for rapid prediction of EGFR overactivation and its application to the rare mutations S768I, S768N, D761N. Comput Struct Biotechnol J 2025; 27:3370-8. doi: 10.1016/j.csbj.2025.07.046 [Crossref] [ Google Scholar]
- Arjama M, Mehnath S, Rajan M, Jeyaraj M. Engineered hyaluronic acid-based smart nanoconjugates for enhanced intracellular drug delivery. J Pharm Sci 2023; 112:1603-14. doi: 10.1016/j.xphs.2021.10.005 [Crossref] [ Google Scholar]
- Qiao Y, Wan J, Zhou L, Ma W, Yang Y, Luo W. Stimuli-responsive nanotherapeutics for precision drug delivery and cancer therapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2019; 11:e1527. doi: 10.1002/wnan.1527 [Crossref] [ Google Scholar]
- Yang Y, Zeng W, Huang P, Zeng X, Mei L. Smart materials for drug delivery and cancer therapy. View (Beijing) 2021; 2:20200042. doi: 10.1002/viw.20200042 [Crossref] [ Google Scholar]
- Irshad S, Siddiqui B, Rehman AU, Farooq RK, Ahmed N. Recent trends and development in targeted delivery of therapeutics through enzyme responsive intelligent nanoplatform. Int J Polym Mater Polym Biomater 2022; 71:403-13. doi: 10.1080/00914037.2020.1848829 [Crossref] [ Google Scholar]
- Sharma S, Chakraborty M, Yadav D, Dhullap A, Singh R, Verma RK. Strategic developments in polymer-functionalized liposomes for targeted colon cancer therapy: an updated review of clinical trial data and future horizons. Biomacromolecules 2024; 25:5650-69. doi: 10.1021/acs.biomac.4c00847 [Crossref] [ Google Scholar]
- Salah Othman R, Zarei S, Rezaei Haghighat H, Afshar Taromi A, Khonakdar HA. Recent advances in smart polymeric micelles for targeted drug delivery. Polym Adv Technol 2025; 36:e70180. doi: 10.1002/pat.70180 [Crossref] [ Google Scholar]
- Li H, Luo Q, Zhang H, Ma X, Gu Z, Gong Q. Nanomedicine embraces cancer radio-immunotherapy: mechanism, design, recent advances, and clinical translation. Chem Soc Rev 2023; 52:47-96. doi: 10.1039/d2cs00437b [Crossref] [ Google Scholar]
- Bhattacharjee K, Prasad BLV. Correction: surface functionalization of inorganic nanoparticles with ligands: a necessary step for their utility. Chem Soc Rev 2023; 52:4121. doi: 10.1039/d3cs90048g [Crossref] [ Google Scholar]
- Singh D. Advancements in hydroxypropyl methylcellulose-polyethylene glycol hybrid excipients and their nanoformulations for controlled and ligand-directed biologic delivery. Naunyn Schmiedebergs Arch Pharmacol 2025. doi: 10.1007/s00210-025-04449-5.
- Esa M, Kaewpaiboon S, Srichana T. Fabrication, biodistribution, and toxicological evaluation of mesoporous silica nanoparticles based on preclinical studies intended for cancer therapy: a review. J Appl Pharm Sci 2025; 15:1-26. doi: 10.7324/japs.2025.220858 [Crossref] [ Google Scholar]
- Patil SR, Adhyapak A, Koli R. Advancing darunavir delivery: nanoformulation strategies and innovations in HIV therapy. Pharm Dev Technol 2025; 30:772-820. doi: 10.1080/10837450.2025.2520624 [Crossref] [ Google Scholar]
- Chang M, Zhang F, Wei T, Zuo T, Guan Y, Lin G. Smart linkers in polymer-drug conjugates for tumor-targeted delivery. J Drug Target 2016; 24:475-91. doi: 10.3109/1061186x.2015.1108324 [Crossref] [ Google Scholar]
- Wu YW, Goubran H, Seghatchian J, Burnouf T. Smart blood cell and microvesicle-based Trojan horse drug delivery: merging expertise in blood transfusion and biomedical engineering in the field of nanomedicine. Transfus Apher Sci 2016; 54:309-18. doi: 10.1016/j.transci.2016.04.013 [Crossref] [ Google Scholar]
- Singh AK, Yadav TP, Pandey B, Gupta V, Singh SP. Engineering nanomaterials for smart drug release: recent advances and challenges. In: Mohapatra SS, Ranjan S, Dasgupta N, Mishra RK, Thomas S, eds. Applications of Targeted Nano Drugs and Delivery Systems. Elsevier. 2019. p. 411-49. doi: 10.1016/b978-0-12-814029-1.00015-6.
- Xu L, Xu S, Xiang T, Liu H, Chen L, Jiang B. Multifunctional building elements for the construction of peptide drug conjugates. Eng Regen 2022; 3:92-109. doi: 10.1016/j.engreg.2022.02.004 [Crossref] [ Google Scholar]
- Lächelt U, Wagner E. Invading target cells: multifunctional polymer conjugates as therapeutic nucleic acid carriers. Front Chem Sci Eng 2011; 5:275-86. doi: 10.1007/s11705-011-1203-z [Crossref] [ Google Scholar]
- Benjeddou A. Smart structures, materials and nano technology in engineering. Int J Smart Nano Mater 2018; 9:85-7. doi: 10.1080/19475411.2018.1463938 [Crossref] [ Google Scholar]
- Omidi Y. Smart multifunctional theranostics: simultaneous diagnosis and therapy of cancer. Bioimpacts 2011; 1:145-7. doi: 10.5681/bi.2011.019 [Crossref] [ Google Scholar]
- Chen YX, Triola G, Waldmann H. Bioorthogonal chemistry for site-specific labeling and surface immobilization of proteins. Acc Chem Res 2011; 44:762-73. doi: 10.1021/ar200046h [Crossref] [ Google Scholar]
- Stayton PS, Ding Z, Hoffman AS. Smart polymer-streptavidin conjugates. In: Niemeyer CM, ed. Bioconjugation Protocols: Strategies and Methods. Totowa, NJ: Humana Press. 2004. p. 37-43. doi: 10.1385/1-59259-813-7:037.
- Leiro V, Parreira P, Freitas SC, Martins MC, Pêgo AP. Conjugation chemistry principles and surface functionalization of nanomaterials. In: Sarmento B, das Neves J, eds. Biomedical Applications of Functionalized Nanomaterials. Elsevier. 2018. p. 35-66. doi: 10.1016/b978-0-323-50878-0.00002-1.
- Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007; 11:217-27. doi: 10.1016/j.ccr.2006.12.017 [Crossref] [ Google Scholar]
- Zhao Z, Bourne PE. Progress with covalent small-molecule kinase inhibitors. Drug Discov Today 2018; 23:727-35. doi: 10.1016/j.drudis.2018.01.035 [Crossref] [ Google Scholar]
- Spada A, Gerber-Lemaire S. Surface functionalization of nanocarriers with anti-EGFR ligands for cancer active targeting. Nanomaterials (Basel) 2025; 15:158. doi: 10.3390/nano15030158 [Crossref] [ Google Scholar]
- Shimoboji T, Ding Z, Stayton PS, Hoffman AS. Mechanistic investigation of smart polymer-protein conjugates. Bioconjug Chem 2001; 12:314-9. doi: 10.1021/bc000107b [Crossref] [ Google Scholar]
- Dahri M, Abolmaali SS, Maleki R, Najafi H, Abedanzadeh M, Tamaddon AM. Nanoscale aggregation of doxorubicin-short peptide conjugates for enzyme-responsive delivery with various MOF carriers: in-silico steps towards smart cancer chemotherapy. Comput Biol Med 2022; 144:105386. doi: 10.1016/j.compbiomed.2022.105386 [Crossref] [ Google Scholar]
- Capek I. Viral nanoparticles, noble metal decorated viruses and their nanoconjugates. Adv Colloid Interface Sci 2015; 222:119-34. doi: 10.1016/j.cis.2014.04.008 [Crossref] [ Google Scholar]
- He H, Wu Y, Chen M, Qi L, He X, Wang K. Acidic extracellular pH-activated allosteric DNA nanodevice for fluorescence imaging of APE1 activity in tumor cells. Anal Chem 2024; 96:18079-85. doi: 10.1021/acs.analchem.4c03934 [Crossref] [ Google Scholar]
- Cheng T, Zhang Y, Liu J, Ding Y, Ou H, Huang F. Ligand-switchable micellar nanocarriers for prolonging circulation time and enhancing targeting efficiency. ACS Appl Mater Interfaces 2018; 10:5296-304. doi: 10.1021/acsami.7b18137 [Crossref] [ Google Scholar]
- Khan N, Ruchika Ruchika, Dhritlahre RK, Saneja A. Recent advances in dual-ligand targeted nanocarriers for cancer therapy. Drug Discov Today 2022; 27:2288-99. doi: 10.1016/j.drudis.2022.04.011 [Crossref] [ Google Scholar]
- Miyazaki T, Nakagawa Y, Cabral H. Strategies for ligand-installed nanocarriers. In: Lau WJ, Faungnawakij K, Piyachomkwan K, Ruktanonchai UR, eds. Handbook of Nanotechnology Applications. Elsevier. 2021. p. 633-55. doi: 10.1016/b978-0-12-821506-7.00024-7.
- Eloy JO, Petrilli R, Lee RJ. Targeting of drug nanocarriers. In: Eloy JO, Abriata JP, Marchetti JM, eds. Nanocarriers for Drug Delivery: Concepts and Applications. Cham: Springer International Publishing. 2021. p. 107-26. doi: 10.1007/978-3-030-63389-9_6.
- Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012; 44:852-60. doi: 10.1038/ng.2330 [Crossref] [ Google Scholar]
- Nasiri R, Sankaranthi A, Pratx G. Organ-on-a-chip systems for modeling tumor and normal tissue microenvironments in radiotherapy research. Trends Biotechnol 2025. doi: 10.1016/j.tibtech.2025.07.002.
- Chaux A. Molecular Mechanisms of Targeted Therapy Resistance in Genitourinary Tumors: A Path to New Therapeutic Horizons. Preprints. June 12, 2025. Available from: https://www.preprints.org/manuscript/202506.1000.
- Mi P, Cabral H, Kataoka K. Ligand-installed nanocarriers toward precision therapy. Adv Mater 2020; 32:e1902604. doi: 10.1002/adma.201902604 [Crossref] [ Google Scholar]
- Song K, Zhang J, Lu S. Progress in allosteric database. In: Zhang J, Nussinov R, eds. Protein Allostery in Drug Discovery. Singapore: Springer. 2019. p. 65-87. doi: 10.1007/978-981-13-8719-7_4.
- Bols PS, Rickhaus M, Tejerina L, Gotfredsen H, Eriksen K, Jirasek M. Allosteric cooperativity and template-directed synthesis with stacked ligands in porphyrin nanorings. J Am Chem Soc 2020; 142:13219-26. doi: 10.1021/jacs.0c06269 [Crossref] [ Google Scholar]
- Guarnera E, Berezovsky IN. Statistical physics of the causality and energetics in allosteric communication. Biophys J 2016; 110:54a. doi: 10.1016/j.bpj.2015.11.354 [Crossref] [ Google Scholar]
- Okafor CD, Ortlund EA. Ligand modulation of allosteric networks in an ancestral steroid receptor. bioRxiv [Preprint]. September 11, 2018. Available from: https://www.biorxiv.org/content/10.1101/414375v1.
- Li N, Binder WH. Click-chemistry for nanoparticle-modification. J Mater Chem 2011; 21:16717-34. doi: 10.1039/c1jm11558h [Crossref] [ Google Scholar]
- von Maltzahn G, Ren Y, Park JH, Min DH, Kotamraju VR, Jayakumar J. In vivo tumor cell targeting with "click" nanoparticles. Bioconjug Chem 2008; 19:1570-8. doi: 10.1021/bc800077y [Crossref] [ Google Scholar]
- Li Y, Dong Y, Shen D, Guo Y, Cao Y, Zhang K. Personalized nanovaccine based on STING-activating nanocarrier for robust cancer immunotherapy. ACS Nano 2025; 19:3226-39. doi: 10.1021/acsnano.4c11014 [Crossref] [ Google Scholar]
- Moura LI, Malfanti A, Matos AI, Peres C, Armiñán A, Duro-Castaño A. Off-the-shelf multivalent nanoconjugate cancer vaccine rescues host immune response against melanoma. Adv Mater 2025; 37:e2417348. doi: 10.1002/adma.202417348 [Crossref] [ Google Scholar]
- Yoon MS, Lee JM, Jo MJ, Kang SJ, Yoo MK, Park SY. Dual-drug delivery systems using hydrogel-nanoparticle composites: recent advances and key applications. Gels 2025; 11:520. doi: 10.3390/gels11070520 [Crossref] [ Google Scholar]
- Yi G, Son J, Yoo J, Park C, Koo H. Application of click chemistry in nanoparticle modification and its targeted delivery. Biomater Res 2018; 22:13. doi: 10.1186/s40824-018-0123-0 [Crossref] [ Google Scholar]
- Elliott EW 3rd, Ginzburg AL, Kennedy ZC, Feng Z, Hutchison JE. Single-step synthesis of small, azide-functionalized gold nanoparticles: versatile, water-dispersible reagents for click chemistry. Langmuir 2017; 33:5796-802. doi: 10.1021/acs.langmuir.7b00632 [Crossref] [ Google Scholar]
- Bai Y, Feng X, Xing H, Xu Y, Kim BK, Baig N. A highly efficient single-chain metal-organic nanoparticle catalyst for alkyne-azide "click" reactions in water and in cells. J Am Chem Soc 2016; 138:11077-80. doi: 10.1021/jacs.6b04477 [Crossref] [ Google Scholar]
- Sun EY, Josephson L, Weissleder R. "Clickable" nanoparticles for targeted imaging. Mol Imaging 2006; 5:122-8. doi: 10.2310/7290.2006.00013 [Crossref] [ Google Scholar]
- Chen Y, Xianyu Y, Wu J, Yin B, Jiang X. Click chemistry-mediated nanosensors for biochemical assays. Theranostics 2016; 6:969-85. doi: 10.7150/thno.14856 [Crossref] [ Google Scholar]
- Thorek DL, Elias DR, Tsourkas A. Comparative analysis of nanoparticle-antibody conjugations: carbodiimide versus click chemistry. Mol Imaging 2009; 8:221-9. doi: 10.2310/7290.2009.00021 [Crossref] [ Google Scholar]
- Dolci M, Toulemon D, Chaffar Z, Bubendorff JL, Tielens F, Calatayud M. Nanoparticle assembling through click chemistry directed by mixed SAMs for magnetic applications. ACS Appl Nano Mater 2018; 2:554-65. doi: 10.1021/acsanm.8b02152 [Crossref] [ Google Scholar]
- Lee SH, Park OK, Kim J, Shin K, Pack CG, Kim K. Deep tumor penetration of drug-loaded nanoparticles by click reaction-assisted immune cell targeting strategy. J Am Chem Soc 2019; 141:13829-40. doi: 10.1021/jacs.9b04621 [Crossref] [ Google Scholar]
- Treerattrakoon K, Chanthima W, Apiwat C, Dharakul T, Bamrungsap S. Oriented conjugation of antibodies against the epithelial cell adhesion molecule on fluorescently doped silica nanoparticles for flow-cytometric determination and in vivo imaging of EpCAM, a biomarker for colorectal cancer. Microchim Acta 2017; 184:1941-50. doi: 10.1007/s00604-017-2211-6 [Crossref] [ Google Scholar]
- Jeong S, Park JY, Cha MG, Chang H, Kim YI, Kim HM. Highly robust and optimized conjugation of antibodies to nanoparticles using quantitatively validated protocols. Nanoscale 2017; 9:2548-55. doi: 10.1039/c6nr04683e [Crossref] [ Google Scholar]
- Byzova NA, Safenkova IV, Slutskaya ES, Zherdev AV, Dzantiev BB. Less is more: a comparison of antibody-gold nanoparticle conjugates of different ratios. Bioconjug Chem 2017; 28:2737-46. doi: 10.1021/acs.bioconjchem.7b00489 [Crossref] [ Google Scholar]
- Moradi N, Muhammadnejad S, Delavari H, Pournoori N, Oghabian MA, Ghafouri H. Bio-conjugation of anti-human CD3 monoclonal antibodies to magnetic nanoparticles by using cyanogen bromide: a potential for cell sorting and noninvasive diagnosis. Int J Biol Macromol 2021; 192:72-81. doi: 10.1016/j.ijbiomac.2021.09.129 [Crossref] [ Google Scholar]
- Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016; 534:129-32. doi: 10.1038/nature17960 [Crossref] [ Google Scholar]
- Yan H, Li Z, Li N, Yan H, Guo X, Wu M. Late course adaptive radiotherapy based on tumor volume reduction decreases gastrointestinal toxicity for abdominal lymph node metastasis of hepatocellular carcinoma. Sci Rep 2025; 15:17477. doi: 10.1038/s41598-025-02363-7 [Crossref] [ Google Scholar]
- Van Zundert I, Bravo M, Deschaume O, Cybulski P, Bartic C, Hofkens J, et al. Versatile and robust method for antibody conjugation to nanoparticles with high targeting efficiency. Pharmaceutics 2021; 13. doi: 10.3390/pharmaceutics13122153.
- Liao Y, Wang Z, Guo M, Yang Y, Chu X, Zhang L. Dual-responsive zwitterionic nanogels with synergistic charge reversal and hypoxia-activated drug release for precise tumor targeting and enhanced chemotherapy. Colloids Surf B Biointerfaces 2025; 254:114839. doi: 10.1016/j.colsurfb.2025.114839 [Crossref] [ Google Scholar]
- Adhikari A, Chen IA. Antibody-nanoparticle conjugates in therapy: combining the best of two worlds. Small 2025; 21:e2409635. doi: 10.1002/smll.202409635 [Crossref] [ Google Scholar]
- Park J, Chariou PL, Steinmetz NF. Site-specific antibody conjugation strategy to functionalize virus-based nanoparticles. Bioconjug Chem 2020; 31:1408-16. doi: 10.1021/acs.bioconjchem.0c00118 [Crossref] [ Google Scholar]
- Johnston MC, Scott CJ. Antibody conjugated nanoparticles as a novel form of antibody drug conjugate chemotherapy. Drug Discov Today Technol 2018; 30:63-9. doi: 10.1016/j.ddtec.2018.10.003 [Crossref] [ Google Scholar]
- Rezaeipoor R, John R, Adie SG, Chaney EJ, Marjanovic M, Oldenburg AL. Fc-directed antibody conjugation of magnetic nanoparticles for enhanced molecular targeting. J Innov Opt Health Sci 2009; 2:387-96. doi: 10.1142/s1793545809000693 [Crossref] [ Google Scholar]
- Juan A, Cimas FJ, Bravo I, Pandiella A, Ocaña A, Alonso-Moreno C. An overview of antibody conjugated polymeric nanoparticles for breast cancer therapy. Pharmaceutics 2020; 12:802. doi: 10.3390/pharmaceutics12090802 [Crossref] [ Google Scholar]
- Escareño N, Hassan N, Kogan MJ, Juárez J, Topete A, Daneri-Navarro A. Microfluidics-assisted conjugation of chitosan-coated polymeric nanoparticles with antibodies: significance in drug release, uptake, and cytotoxicity in breast cancer cells. J Colloid Interface Sci 2021; 591:440-50. doi: 10.1016/j.jcis.2021.02.031 [Crossref] [ Google Scholar]